
论文地址:Democratizing protein language model training, sharing and collaboration
SaProtHub:训练微调共享saprot平台
Abstract
训练和部署大规模蛋白质语言模型(PLM)通常需要深厚的机器学习专业知识——这对该领域之外的研究人员来说是一个障碍 。SaprotHub 通过提供一个直观的平台克服了这一挑战,该平台促进了模型的训练和预测,以及存储和共享 。提供了基于 Google Colab 构建的 ColabSaprot 框架,它有潜力为数百个蛋白质训练和预测应用提供动力,使研究人员能够协作构建和共享定制模型 。
1 Main
引言与背景
蛋白质几乎是所有生物过程的基础,并且是医学和生物技术的核心。尽管具有这种中心地位,破译蛋白质结构和功能仍然是一个艰巨的挑战。这一格局最近被两项突破所改变:AlphaFold2 的成功通过以实验级准确性预测结构,开创了结构生物学的新纪元;与此同时,大规模蛋白质语言模型 (PLM) 正在推动功能预测领域前所未有的进步。
这一进展是由一套强大的 PLM 推动的,它们在各种任务中都表现出了卓越的功效。然而,对于没有广泛机器学习 (ML) 专业知识的研究人员来说,利用这些先进模型存在显著的技术障碍。这些挑战贯穿整个工作流程,从模型选择、数据预处理,到训练和评估拥有数十亿参数的模型。这种复杂性创造了一个关键障碍,阻碍了那些最能从中受益的研究人员的广泛采用和创新。
ColabFold 通过在 Google Colab 上部署 AlphaFold2,解决了结构预测方面类似的“可及性”障碍,有效地普及了其使用。尽管取得了这一成功,一个关键的空白仍然存在:ColabFold 不支持更复杂的、用于功能预测的自定义模型训练任务。
解决方案:ColabSaprot, SaprotHub 与 OPMC
为了弥合这一差距,作者引入了 ColabSaprot 和 SaprotHub,这是一个专为蛋白质功能预测设计的平台。ColabSaprot 建立在 Google Colab 之上,使没有 ML 专业知识的研究人员能够通过直观的界面训练他们自己特定任务的 PLM。至关重要的是,该平台支持广泛的预测任务,确保其效用远远超出了单任务应用。
作为这个用户友好平台的补充,介绍了开放蛋白质建模联盟 (OPMC),这是一项旨在培养社区驱动的蛋白质语言建模协作生态系统的倡议。OPMC 框架使研究人员能够分享他们定制的模型,微调同行贡献的现有模型,或将它们直接应用于自己的研究。这创造了一个共享、优化和应用的良性循环,加速了该领域的集体进步。作为第一个与 OPMC 集成的平台,SaprotHub 代表了实现这一以社区为中心的人工智能 (AI) 发展愿景的第一步。
三项核心贡献
这项工作包含三个关键贡献:
- 一个名为 Saprot 的基础 PLM (图 1a, b)。
- ColabSaprot (图 2a–c),它通过适配器学习技术 (adapter learning technique),使得在 Colab 平台上能够轻松地训练(或微调)和推理 Saprot。
- SaprotHub,一个用于存储、共享、搜索和协作开发微调后 Saprot 模型的社区存储库 (图 2d)。
通过整合先进的 PLM、基于 Colab 的云计算和基于适配器的微调技术,它解决了几个关键挑战——即共享和集体使用大型 PLM 的困难、持续学习中参数“灾难性遗忘” (catastrophic forgetting) 的风险,以及在共享结果时保护专有生物数据的需求。
1. Saprot:结构感知的基础模型
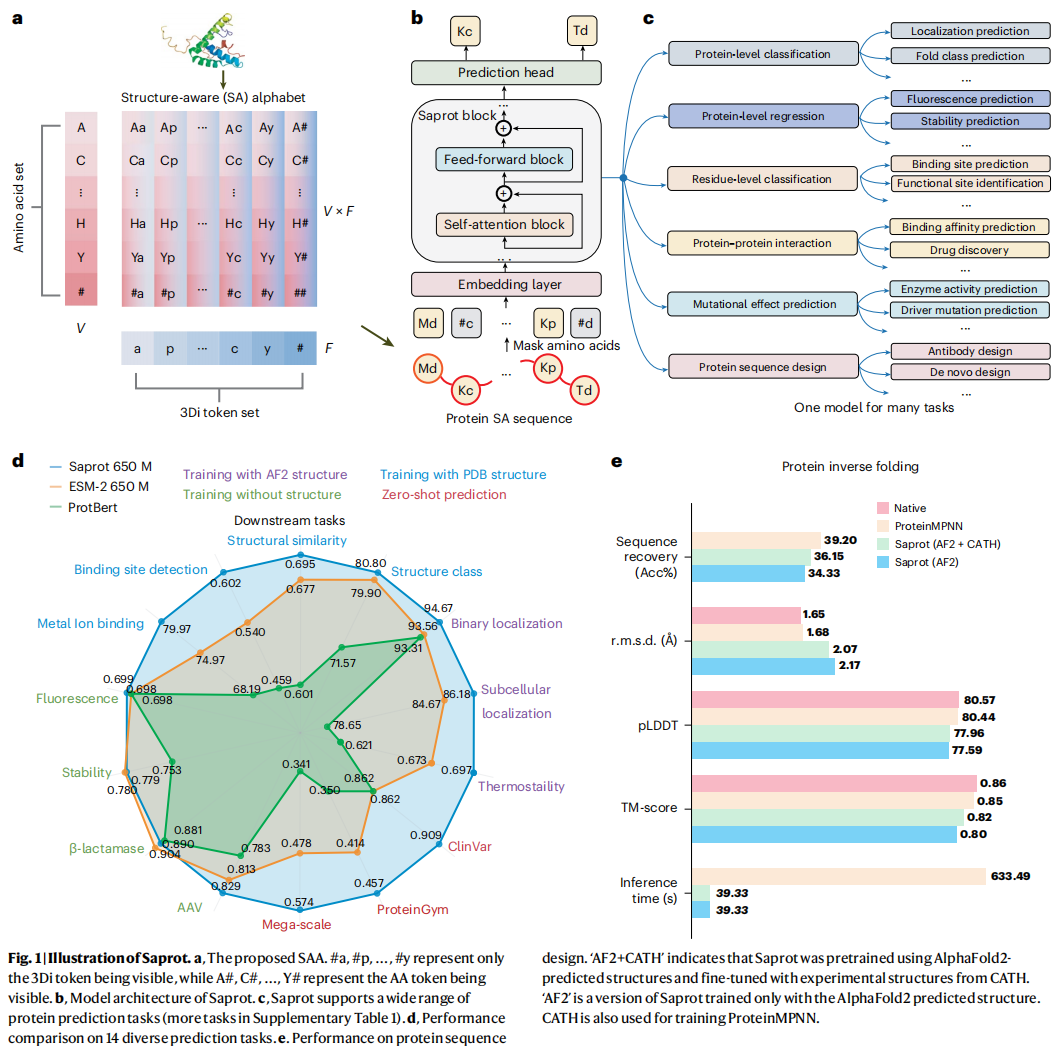
首先开发了 Saprot,一个前沿的大规模 PLM,它构成了 ColabSaprot 和 SaprotHub 的基础。Saprot 引入了一种新颖的蛋白质字母表和表示方法,不同于传统的氨基酸 (AA) 序列或显式的三维 (3D) 坐标结构。
这个字母表是结构感知 (Structure-Aware, SA) 的,每个“字母”都同时编码 AA 类型和蛋白质的局部几何结构。形式上,SA 字母表 (SAA) 被定义为 $SAA = \mathcal{V} \times \mathcal{F}$,代表 $\mathcal{V}$ (20 种 AA) 和 $\mathcal{F}$ (20 种结构 (3Di) 字母) 的笛卡尔积 (图 1a)。这些 3Di 令牌是通过 Foldseek 离散化从蛋白质 3D 结构中派生出来的。SAA 包含了 AA(大写字母)和 3Di 令牌(小写字母)的所有可能组合 (例如 Aa, Ap, Ac, …, Ya, Yp, Yy),允许蛋白质被表示为捕获一级和三级结构的 SA 令牌序列 (图 1b 和方法)。
尽管 SAA 很简洁,但它成功解决了在大型 AlphaFold2 生成的原子结构上训练时的可扩展性和过拟合等关键挑战。“AA + 结构令牌”序列 (图 1b) 用于蛋白质表示的方法已在后续研究中获得越来越多的关注,成为蛋白质表示的一个有前途的范式。
Saprot 模型使用双向 Transformer 架构 (图 1b)。它经过预训练,用于重建 SA 令牌序列中某些被部分掩码的令牌。该模型在 4000 万个蛋白质 SA 序列上从头开始训练,这些序列是从 AlphaFold 的 2.14 亿个蛋白质中以 50% 的一致性过滤出来的。Saprot 提供三种大小:35M、650M 和 1.3B(M,百万;B,十亿)。650M 模型(用于大多数评估)使用了 64 个 NVIDIA A100 80-GB GPU 训练了 3 个月,其计算资源与 ESM-2 (650M) 相当。
预训练后,Saprot 已成为一个通用的基础 PLM,在各种蛋白质预测任务中表现出色,例如各种监督训练任务(蛋白质级别或残基级别的回归与分类)、零样本 (zero-shot) 突变效应预测和蛋白质序列设计 (图 1c, d)。
图 1d 显示了 Saprot 在 14 种不同蛋白质预测任务上相比两个著名 PLM:ESM-2 和 ProtBert 的卓越性能。在有蛋白质结构信息可用的任务中(用紫色、蓝色和红色表示),Saprot 始终超越这两个模型。即使在没有结构数据的场景中(绿色任务),Saprot 在微调设置下也保持了与 ESM-2 相当的竞争力。
值得注意的是,Saprot 在三个零样本突变效应预测任务上显著优于 ESM-2:Mega-scale (0.574 vs 0.478)、ProteinGym (0.457 vs 0.414) 和 ClinVar (0.909 vs 0.862)。此外,尽管 Saprot 是用掩码语言建模目标训练的(并非专门为生成任务优化),但它在蛋白质序列设计方面表现有效,同时推理速度比 ProteinMPNN 快 16 倍 (图 1e)。最近的研究还证明了 Saprot 在蛋白质工程、从头设计、适应度和稳定性预测、分子理解、快速敏感的结构搜索以及药物-靶标相互作用预测等广泛应用中的有效性。这种跨多项任务的多功能性支持了 SaprotHub 的社区协作愿景。
2. ColabSaprot:易用的训练与预测平台
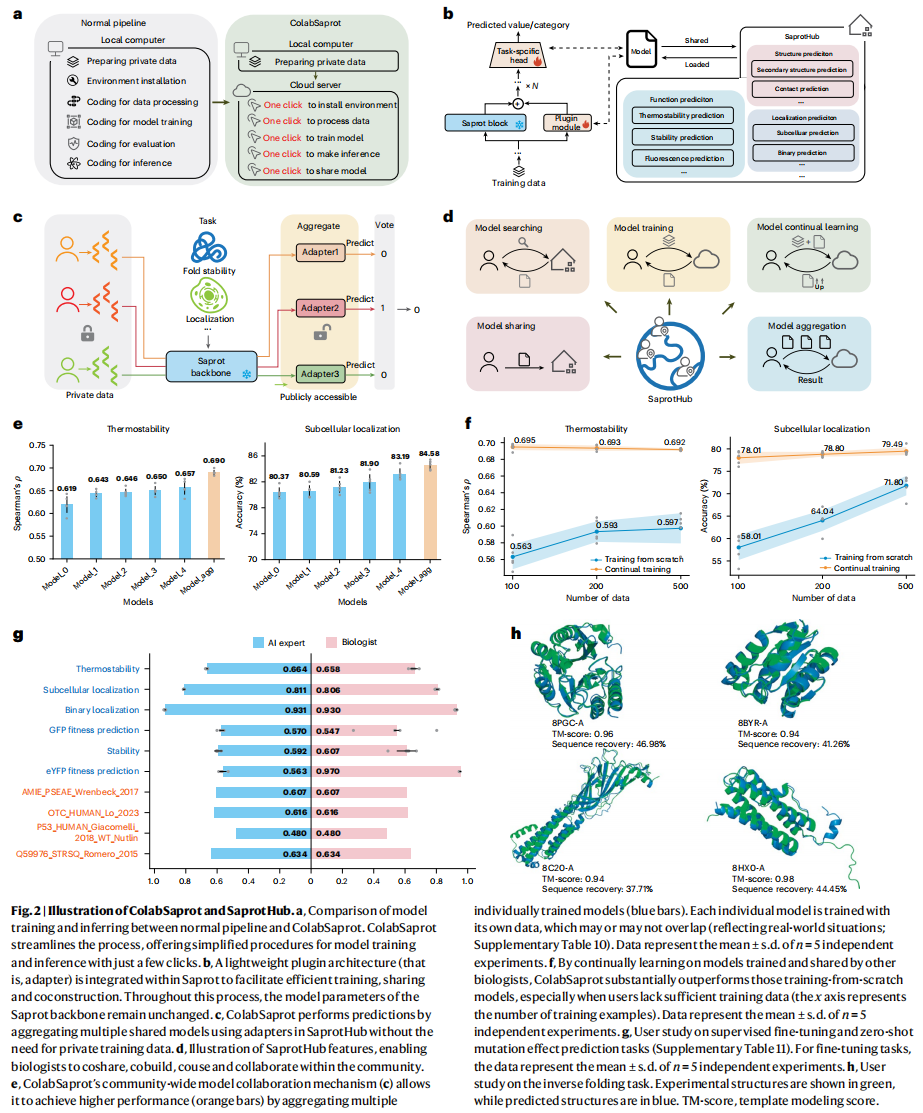
接着通过将 Saprot 集成到 Google Colab 的基础架构中,开发了 ColabSaprot 平台,以支持 PLM 训练和预测。ColabSaprot 能够无缝部署和执行各种任务特定的已训练 Saprot 模型,无需进行环境设置和代码调试。它还允许研究人员只需点击几下即可启动训练会话 (图 2a)。ColabSaprot 旨在容纳原始 Saprot 框架内的所有任务,支持零样本突变效应预测(实现单点、多点和全序列单点饱和突变)和蛋白质序列设计(可根据给定的骨架结构生成从头序列)等任务的直接预测。
对于监督训练任务(这项工作的特别焦点),用户可以使用自己的实验数据微调 ColabSaprot。在这里通过在 ColabSaprot 中集成轻量级适配器网络 (lightweight adapter networks),实现了一种参数高效微调 (PEFT) 技术 (图 2b)。
在训练期间,只有适配器参数被更新,而 Saprot 骨干参数保持不变,这实现了与微调所有 Saprot 参数相当的准确性。这种设计不仅提高了学习效率,还建立了一个协作和集中的框架,使生物学家能够在研究社区内(特别是通过云环境)微调特定任务的 Saprot (图 2c–f)。
借助适配器和 ColabSaprot 界面,研究人员可以通过加载或上传适配器网络(而非完整的预训练模型)来轻松地在 SaprotHub 中存储和交换他们重新训练的模型。由于适配器网络包含的参数要少得多(约为整个 Saprot 模型的 1%),这种方法极大地减少了 SaprotHub 的存储、通信和管理负担,使模型的“可及性、共建、共用和共享”成为可能 (图 2b–d)。
此外,开发了几个关键功能来简化研究人员的工作流程。ColabSaprot 界面支持序列和结构输入,提供自动化数据集处理能力,包括高效的大文件上传机制、带损失可视化的实时训练监控、最佳检查点的自动保存和评估、断点训练恢复,以及众多安全检查以最小化用户错误。为了提高 GPU 内存效率,通过梯度累积实现了自适应批量大小 (adaptive batch sizing)。该平台还包括可自定义的设置,允许研究人员修改代码和调整训练参数以满足特定的研究需求。
3. SaprotHub:协作与共享的社区
最后,开发了 SaprotHub,一个集成了搜索引擎的平台,为生物学界提供了共享和协作开发同行再训练 (peer-retrained) Saprot 模型的中央平台 (图 2d)。通过 ColabSaprot 界面实现了模型存储、模型共享、模型搜索、模型持续学习和多模型聚合等功能。
具体来说,SaprotHub 提供了三个关键优势:
- 安全共享:研究人员可以在 SaprotHub 上分享他们训练好的模型,而不必担心私有数据的泄露。这有效地促进了知识传播,并有潜力建立协作研究的新范式 (图 2c)。
- 持续学习:研究人员可以利用 SaprotHub 上同行贡献的共享模型,在自己的数据集上执行持续训练。这种方法在数据有限的场景中特别有利,因为从一个更好的预训练模型微调通常能带来更优的预测性能 (图 2f)。此外,这些微调后的模型还可以通过一键上传适配器网络贡献回中心,形成协作生态。
- 模型聚合:随着 SaprotHub 社区的扩展,它将为各种蛋白质预测任务积累多个模型。实现了一种模型聚合机制 (图 2c),使 用户能够通过整合多个现有模型来增强预测性能 (图 2e)。
湿实验验证与用户研究
Saprot 和 SaprotHub 已经吸引了社区关注,并通过多次湿实验验证证明了其有用性。
- 一家商业生物公司的 T.Z. 团队使用 ColabSaprot 对一种木聚糖酶 (xylanase) 进行零样本单点突变预测,实验验证的 20 个变体中,13 个表现出增强的酶活性,其中 R59S 活性提高了 2.55 倍。
- X.C. 实验室使用 ColabSaprot 对 TDG 变体进行零样本单点突变预测,在 HeLa 细胞中验证的 20 个预测突变中有 17 个显示出增强的编辑效率,其中三个替换实现了近两倍的效率。
- 另一位 OPMC 成员微调 Saprot 预测更亮的 GFP 变体,实验验证显示,排名前 9 位的双位点变体中有 7 个表现出增强的荧光,其中一个变体的荧光强度达到了野生型的八倍以上。
- J. Zheng 实验室共享了一个 eYFP 荧光预测模型,该模型在 100,000 个实验验证过的变体上训练,在独立测试集上实现了 0.94 的斯皮尔曼相关性 (ρ),显示了接近实验级的准确性。
- 作者最近收到了更多来自社区研究人员使用 ColabSaprot 获得积极湿实验结果的反馈。
还进行了一项用户研究,招募了 12 名没有 ML 背景的生物学研究人员,并将他们的表现与一名 AI 专家进行比较。结果表明,借助 ColabSaprot 和 SaprotHub,生物学研究人员可以训练和使用最先进的 PLM,其性能与 AI 专家相当 (图 2g, h)。
值得注意的是,在某些场景中——例如 eYFP 适应度预测任务 (图 2g)——生物学家利用 SaprotHub 上的预有模型所取得的预测准确性甚至高于 AI 专家。这种更高的性能源于这样一个事实:这些共享模型是在更大或更高质量的数据集上训练的,这突显了科学界内部模型共享的潜力——这也是本文的一个关键论点。
结论与愿景
ColabSaprot 和 SaprotHub 使生物学研究人员(即使没有广泛的 AI 专业知识)能够为多样化的预测任务训练和共享复杂的 PLM。该平台赋能更广泛的蛋白质研究社区贡献和交换 PLM,通过同行训练的模型促进协作研究和知识共享。
已经将 Saprot 和 ColabSaprot 开源,为其他 PLM 开发自己的模型中心提供了框架。重要的是,ColabSaprot 和 SaprotHub 只是这一演进的第一步; OPMC 成员已经扩展了此生态系统,集成了更多前沿的 PLM,包括 ProTrek、ESM-2、ESM-1b、ProtBert 和 ProtT5,从而在全球范围内普及了生物学家对多样化 PLM 的访问。
这种社区范围的参与方法与 OPMC 的愿景相一致,目标是通过 SaprotHub 激励和促进开放 PLM 的合作共建。将 SaprotHub 视为 OPMC 的催化剂,推动该领域的创新和协作。
2 Methods
1. 构建 SA 蛋白质序列 (Constructing SA protein sequence)
SA 词汇表 (SA vocabulary) 同时包含了残基和结构信息。给定一个蛋白质 $P$,其一级序列表示为 $(S_{1}, S_{2}, ..., S_{n})$,其中 $s_{i}$ 是第 $i$ 位的残基。研究者使用 Foldseek 这款快速准确的蛋白质结构比对工具,将蛋白质 3D 结构编码为离散的、类似残基的结构令牌。这产生了一个结构字母表 $\mathcal{F}$,其中蛋白质 $P$ 被表示为序列 $(f_{1}, f_{2}, ..., f_{n})$,其中 $f_{j} \in \mathcal{F}$ 是第 $j$ 个残基位点的 3Di 令牌。
研究者遵循 Foldseek 的默认配置,将 $\mathcal{F}$ 的大小(即 $m$)设为 20。然后,他们将每个残基位点的残基令牌和结构令牌结合起来,生成一个新的 SA 蛋白质序列 $P=(s_{1}f_{1}, s_{2}f_{2}, ..., s_{n}f_{n})$,其中 $s_{i}f_{i} \in \mathcal{V} \times \mathcal{F}$ 是所谓的 SA 令牌,自然地融合了残基和几何构象信息。
SA 令牌蛋白质序列随后可以被送入标准的 Transformer 编码器作为基本输入。研究者还为残基和结构字母表引入了掩码信号 ‘#’,这导致了 ‘$s_{i}\#$’ 和 ‘$\#f_{i}$’ 两种形式,分别表示仅残基信息可用或仅结构信息可用。SA 词汇表的大小因此为 $21 \times 21 = 441$。
2. Saprot 的模型架构和预训练 (Model architecture and pretraining of Saprot)
Saprot 遵循与 ESM-2 相同的网络架构和参数大小,其灵感来自 BERT 模型。主要区别在于嵌入层:Saprot 纳入了 441 个 SA 令牌,以取代传统的 20 个 AA 令牌。这种几乎相同的架构使得与 ESM 模型的性能比较变得直接。
Saprot 使用与 ESM-2 和 BERT 相同的掩码语言建模 (MLM) 目标进行预训练。对于一个蛋白质序列 $P=(s_{1}f_{1}, s_{2}f_{2}, ..., s_{n}f_{n})$,输入和输出可以表示为:输入 $(s_{1}f_{1}, ..., \#f_{i}, ..., s_{n}f_{n}) \rightarrow$ 输出 $s_{i}f_{i}$ (图 1b) 。考虑到 AlphaFold2 预测结构中的 3Di 令牌在某些区域可能不准确,训练时会使得 ‘$\#f_{i}$’ 可见,以减少模型对其预测的依赖。
AlphaFold2 (AF2) 的预测包含 pLDDT 置信度分数。因此,研究者对低置信度分数区域(pLDDT 低于 70)实施了专门处理。在预训练期间,当这些区域被选中进行 MLM 预测时,使用 ‘$s_{i}\#$’ 令牌作为预测目标,同时用 ‘##’ 令牌掩码输入 SA 序列。当这些低置信度区域未被选中进行 MLM 预测时,在输入中使用 ‘$s_{i}\#$’ 令牌,确保模型仅依赖残基上下文而非不准确的结构信息。在下游任务中,也应用了相同的处理方式,pLDDT 低于 70 的区域用 ‘$s_{i}\#$’ 令牌表示。
Saprot 35M 和 650M 进行了如上所述的典型从头预训练。相比之下,Saprot 1.3B 使用了一种高效的训练策略,通过架构上组合两个相同的 33 层 Saprot 650M 模型来实现。初始化过程涉及复制预训练 650M 模型的参数,以填充 1.3B 架构的低层(1-33层)和高层(34-66层)。初始化后,Saprot 1.3B 遵循与 35M 和 650M 模型相同的训练协议,例外之处在于训练批次中 30% 的蛋白质序列被转换为纯 AA 序列格式 $(s_{1}\#, s_{2}\#, ..., s_{n}\#)$,以增强模型处理无可用结构信息蛋白质的能力。
3. 处理预训练数据集 (Processing pretraining dataset)
研究者遵循 ESM-2 中概述的程序来生成序列一致性过滤的蛋白质数据。随后,他们通过 AlphaFold DB 网站获取了所有 AF2 结构。在 AlphaFold DB 中没有结构的蛋白质被移除。这个过程产生了约 4000 万个结构的集合。研究者使用 Foldseek 将这些结构编码为 3Di 令牌,随后通过组合残基和 3Di 令牌来构建 SA 序列。这些数据集被用于训练所有三个版本的 Saprot 模型。
4. 预训练超参数 (Hyperparamters for pretraining)
遵循 ESM-2 和 BERT,训练期间,每批中 15% 的 SA 令牌被掩码。在 80% 的情况下,SA 令牌 $s_{i}f_{i}$ 被替换为 $\#f_{i}$ 令牌;10% 的情况下被替换为随机选择的令牌;另外 10% 保持不变。
优化方面,使用了 AdamW 优化器,设置 $\beta_{1}=0.9$,$\beta_{2}=0.98$(译者注:原文Methods章节中未提 $\beta_{2}$,但在第9页的 fine-tuning 部分提到了 $\beta_{2}=0.98$),并使用了 0.01 的 $L_{2}$ 权重衰减。学习率在前 2,000 步内从 0 逐步增加到 $4 \times 10^{-4}$,并在 150,000 步到 150 万步期间线性降低到 $4 \times 10^{-5}$。总训练阶段持续约 300 万步。序列被截断为最大 1,024 个令牌,批量大小为 512 个序列。训练使用了混合精度。
5. 基线模型描述 (Descriptions of baseline models)
Saprot 与几个著名的 PLM 进行了比较。
- 监督学习任务:比较了 ESM-2 (650M)、ProtBert (BFD 版)、MIF-ST、GearNet 和 ESM-3 40。ESM-2 (650M) 是主要的比较基线,因为其模型架构、大小和训练方法与 Saprot 相似。
- 零样本突变效应预测:比较了 ESM-2 (包括 15B 版本)、ProtBert、ESM-1v、Tranception L (无 MSA 检索)、MSA Transformer、EVE 和 ESM-3。
- 蛋白质反向折叠:比较了 ProteinMPNN。
6. 零样本突变效应预测任务的公式 (The formula for the zero-shot mutation effect prediction task)
以往的 PLM (如 ESM) 使用突变位置的对数几率比 (log odds ratio) 来预测突变效应。公式为:
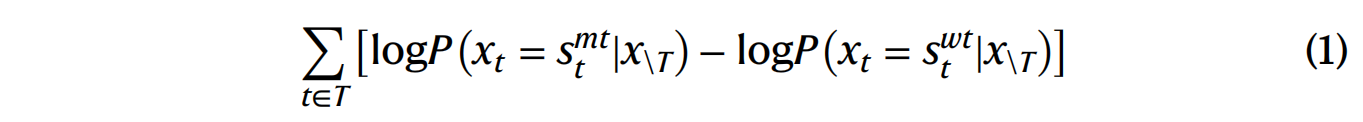
其中 $T$ 代表所有突变,$s_t$ 是突变型 (mt) 和野生型 (wt) 序列的残基类型。
研究者稍微修改了该公式以适应 Saprot 中的 SAA,其中分配给每个残基的概率对应于包含该特定残基类型的所有令牌的概率总和。公式如下:
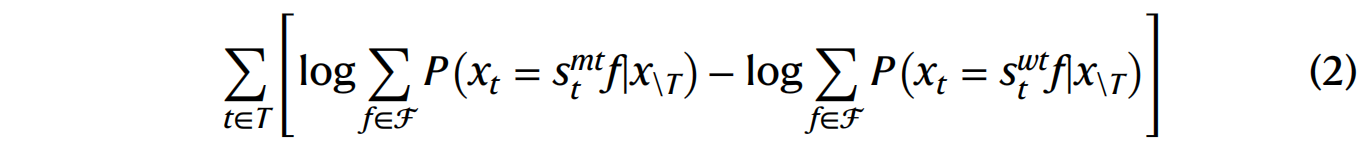
其中 $f \in \mathcal{F}$ 是 3Di 令牌,$s_t f$ 是 SAA 中的 SA 令牌。
7. 零样本突变效应预测任务和数据集 (Zero-shot mutational effect prediction tasks and datasets)
- ProteinGym:一个包含广泛深度突变扫描分析的数据集。评估在 ProteinGym 的替换分支上进行,使用其提供的蛋白质结构,并采用斯皮尔曼等级相关性 (Spearman’s rank correlation) 作为评估指标。
- ClinVar:一个包含人类遗传变异及其临床重要性信息的公共存储库。分析仅限于可靠性评级为“一星”或更高的数据,并排除了长度超过 1,024 个残基的蛋白质。评估使用 ROC 曲线下面积 (AUC) 。
- Mega-scale:使用互补 DNA 展示蛋白水解技术测量蛋白质域的热力学折叠稳定性。该数据集也使用斯皮尔曼等级相关性作为评估指标。
对于每个突变数据集,研究者都为所有变体提供了野生型结构,因为 AF2 不能可靠地区分单点替换引起的结构变化。
8. 监督微调任务和数据集 (Supervised fine-tuning tasks and datasets)
- 使用 AF2 结构微调 Saprot:包括来自 FLIP 的热稳定性 (Thermostability) 任务和来自 DeepLoc 的定位预测 (localization prediction) 任务(包含十类亚细胞位置的多类分类和两类位置的二元分类)。
- 使用 PDB 结构微调 Saprot:包括金属离子结合 (metal-ion-binding) 任务和一系列来自 ProteinShake 的任务(结构类别预测、结构相似性预测和结合位点检测)。
- 无结构微调 Saprot:当结构数据不可用时,在 SA 序列中掩码 3Di 令牌。评估了来自 TAPE 的荧光 (fluorescence) 和稳定性 (stability) 预测、来自 FLIP 的 AAV 数据集以及来自 PEER 的 $\beta$-内酰胺酶 ($\beta$-lactamase) 景观预测。
9. 数据划分 (Data split)
现有文献通常基于蛋白质序列一致性来划分数据集。然而,ProteinShake 基准研究指出,蛋白质结构比序列更保守,因此基于结构的划分(structure-based splitting)能对模型泛化能力提供更严格的评估。
受此启发,研究者在大多数评估中采用了基于结构的划分标准。该标准使用 ProteinShake 中提出的 LDDT 进行量化。具体来说,对于包含蛋白质结构的数据集,使用了 ProteinShake 推荐的默认 70% LDDT 阈值。对于仅有序列数据的任务,保留了原始文献提供的划分。
10. 蛋白质反向折叠 (Protein inverse folding)
为了将 Saprot 用于反向折叠,研究者首先将蛋白质骨架编码为 3Di 令牌 $(f_{1}, f_{2}, ..., f_{n})$ 。然后,他们掩码 SA 令牌的所有残基部分,形成一个 SA 序列 $(\#f_{1}, \#f_{2}, ..., \#f_{n})$ 。该序列被输入 Saprot 以预测所有位置的残基分布。与 ProteinMPNN 的自回归生成方式不同,Saprot 能够通过一次前向传播同时预测所有残基。
研究者评估了两个 Saprot 变体:一个使用 AF2 预测结构进行预训练,另一个在 CATH 数据集上进一步微调。CATH 数据库(也用于训练 ProteinMPNN)被划分为 80:10:10 的训练、验证和测试集。
11. 微调任务的超参数 (Hyperparameters for fine-tuning tasks)
微调期间使用 AdamW 优化器,设置 $\beta_{1}=0.9$,$\beta_{2}=0.98$,以及 0.01 的 $L_{2}$ 权重衰减。所有数据集的批量大小均为 64。对于有 AF2 结构和 PDB 结构的任务,最优学习率设为 $5 \times 10^{-5}$;对于无结构的任务,设为 $1 \times 10^{-5}$。
12. 适配器学习 (Adapter learning)
适配器学习技术最近已被蛋白质研究采用。本文将其与 Google Colab 集成,创建了一个平台,通过 SaprotHub 实现模型微调、共享、持续再训练和协作。
在多种适配器类型(如 Houlsby, Pfeiffer, Compacter, LoRA)中,研究者选择了 LoRA,因为它能以更少的参数提供相当的结果。通过在冻结骨干网络的同时,将可学习的低秩矩阵集成到每个 Saprot 块中,LoRA 实现了参数高效的微调和模型共享。
13. 社区范围的协作 (Community-wide collaboration)
为了评估协作的优势:
- 模型聚合 (Model aggregation) (图 2e):研究者将训练数据随机划分为五个子集,并分别训练一个模型(模拟不同研究者共享的模型)。对于回归任务(如热稳定性),最终预测是所有模型输出的平均值。对于分类任务(如亚细胞定位),采用多数投票。
- 模型持续学习 (Model continue learning) (图 2f):研究者随机采样 100、200 和 500 个训练实例(模拟研究者的私有数据)。他们使用两个基础模型进行微调(仅更新适配器参数):官方 Saprot 模型(蓝色)和来自 SaprotHub 的共享模型(橙色)。结果表明,从现有的、训练良好的模型(橙色)继续训练,性能显著优于从头开始训练(蓝色)。
14. ColabSaprot 笔记本 (ColabSaprot notebooks)
ColabSaprot 由三个关键组件构成:
- 模型训练:使研究人员能够快速配置环境、处理数据和高效微调 Saprot。提供了四种基础模型(Saprot 35M, 650M, 自定义微调模型, 社区共享模型)以及高级超参数定制选项。
- 预测能力:支持广泛的预测任务(蛋白质级别、残基级别、蛋白质间相互作用、零样本突变效应和蛋白质设计),可使用社区共享模型或本地微调模型。ColabSaprot 还通过集成方法(聚合) 来增强预测准确性。
- 模型共享、搜索和协作:研究人员可以将其模型权重(特别是适配器权重)贡献给 SaprotHub 社区存储库。通过 SaprotHub 的专用搜索引擎,研究人员可以定位和使用相关模型,用于持续学习、直接应用或模型聚合。
15. 用户研究 (User study)
研究者通过八项蛋白质预测任务对 ColabSaprot-v1 进行了评估,招募了 12 名生物学背景但没有编码或 ML 项目经验的参与者。评估涵盖了监督微调、零样本突变效应预测和蛋白质反向折叠任务。
作为对比,研究者聘请了一名 AI 专家(一名专攻 ML 的三年级博士生)使用 GitHub 上的 Saprot 代码库执行相同任务。
12 名参与者被分配了任务(例如,10 人分为两组,每组 5 人,处理总共 6 个监督微调任务)。监督微调任务使用了五个公共数据集和一个专有的 eYFP 适应度预测数据集。为了减少训练时间,训练集被随机采样至 1,000 个样本。
在 eYFP 任务中,生物学参与者利用了 SaprotHub 上的预训练模型进行持续学习,而 AI 专家则使用了基础的 Saprot 650M 模型。
16. 木聚糖酶的实验验证 (Experimental validation on the xylanase)
该验证包含多个步骤:
- 使用 AlphaFold3 获取 Mth 蛋白 (XP_069217686.1) 的结构。
- 使用 Foldseek 或 ColabSaprot 获取 SA 序列。
- 使用 ColabSaprot (650M) 对整个 SA 序列进行零样本(单点)突变效应预测。
- 选择了排名前 20 的变体(排除了位于信号肽区域的 A15G 和 I13P)。
- 构建突变体:在 P. pastoris (毕赤酵母) 中进行基因合成、定点诱变、转化和筛选。
- 酶活性测定:在 BMMY 培养基中诱导表达 xylanase,并进行 fed-batch 发酵。使用 3,5-二硝基水杨酸法 (DNS法) 测定木聚糖酶活性,并评估了最适温度和热稳定性。
17. GFP 变体的实验验证 (Experimental validation on GFP variants)
该任务的目标是作为 2024 年蛋白质工程关键评估 (CAPE) 竞赛的一部分,设计更亮的 avGFP 变体 。
- 数据准备:CAPE 组织者提供了 140,000 个 GFP 变体的数据集及其适应度分数和四种野生型 GFP 结构 108。OPMC 成员使用 Foldseek 生成 3Di 令牌,并转换为 SA 令牌。
- 模型微调:使用 Saprot (35M) 模型进行全参数微调。
- 变体预测和验证:通过随机诱变生成了 500 万个 avGFP 双位点突变体库。使用微调后的 Saprot 模型预测其适应度分数。排名前九的变体被选中进行实验验证。
- 实验验证:在深圳合成生物学基础设施的机器人生物铸造厂进行。包括在 E. coli BL21(DE3) 细胞中表达,IPTG 诱导,以及测量
$OD_{600}$和 GFP 荧光(激发 488nm,发射 520nm)。
18. TDG 变体的实验验证 (Experimental validation of the TDG variants)
X.C. 实验室使用了 ColabSaprot v1 (650M) 对 TDG 进行零样本单点突变效应预测。他们仅输入了 AA 序列(所有结构令牌均被掩码)。实验验证了排名前 20 的变体。这些变体被克隆到 PCMV 质粒中,并融合到 SpCas9(D10A) 蛋白上(作为 TSBE2)。实验验证过程如先前所述 。