
GeoStab:PLM+GAT+排序学习预测稳定性突变
Abstract
提出了什么:论文提出了一个名为 GeoStab-suite 的模型套件,包含三个基于几何学习的模型:GeoFitness、GeoDDG 和 GeoDTm 。它们分别用于预测蛋白质突变后的适应性分数(fitness score)
$\Delta\Delta G$(自由能变化)和 $\Delta T_{m}$(熔解温度变化)。
如何解决数据难题:
- GeoFitness 使用一种专门的损失函数,使其能利用深度突变扫描数据库中大量、标签不统一(multi-labeled)的适应性数据进行监督式训练 。
- GeoDDG 和 GeoDTm 则利用 GeoFitness 的编码器作为预训练模块,以克服自身训练数据不足的挑战 。
策略效果如何:这种预训练策略,再结合数据扩充,显著提升了模型的性能和泛化能力 。
最终成果:在基准测试中,GeoDDG 和 GeoDTm 的性能(以斯皮尔曼相关系数衡量)分别比其他顶尖方法高出至少30%和70% 。
1 Introduction
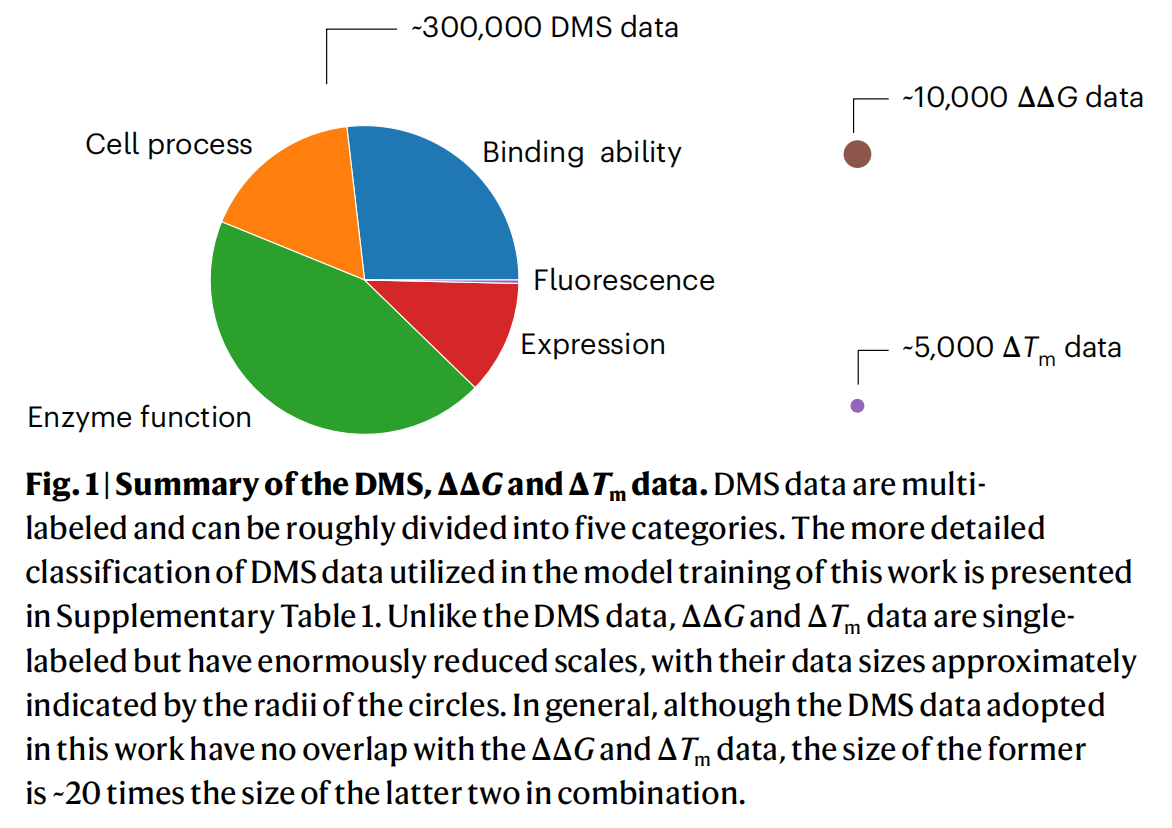
准确预测蛋白质突变效应在蛋白质工程与设计中至关重要 。蛋白质的适应性(fitness)被定义为蛋白质执行特定功能的能力,但在不同的实验案例中,它通常由不同的指标来量化(例如,酶活性、肽结合亲和力以及蛋白质稳定性)。蛋白质设计与工程的主要目标之一就是提高蛋白质的适应性,从而增强其在生物技术和生物制药过程中的性能 。在各种蛋白质适应性指标中,蛋白质稳定性引起了极大的关注,通常由两个度量标准来评估,即 ΔG 和 T_m 。
ΔG 指的是室温下的去折叠自由能变化,描述了蛋白质的热力学稳定性;而 T_m 代表蛋白质的熔解温度,反映了蛋白质在温度波动下维持其折叠状态的能力 。理解蛋白质稳定性不仅有助于获得在严苛生物加工过程或生产条件下能持续保持活性的工程蛋白,也有助于在生物医学领域阐明人类遗传变异在多种疾病病因中的作用 。
蛋白质适应性的预测方法可以基于深度突变扫描(DMS)数据库进行开发和优化 。该数据库包含了超过3,000,000条关于蛋白质突变后适应性的数据 。然而,DMS数据的“多标签”性质阻碍了训练一个统一的预测模型 。现有的模型主要采用两种策略来规避这一障碍 。一种策略,如ECNet和SESNet所应用,是基于每种适应性定义来训练一个特定的模型 。因此,在进行实际预测之前,需要额外的、与目标蛋白属性具有相同含义的适应性数据来重新训练这些模型 。这种策略性能令人满意,但牺牲了模型的易用性 。另一种策略是采用掩码语言建模目标来训练模型,如 RFjoint、MSA Transformer及ESM系列模型,这种方法完全绕过了对任何适应性数据的需求 。该策略可以产生用于快速预测的统一模型,但代价是模型性能的损失 。目前,仍然缺乏一种能够生成性能令人满意的统一适应性预测模型的稳健方法 。
与多标签的适应性数据不同,蛋白质突变引起的稳定性变化由两个明确的度量标准 ΔΔG 和 ΔT_m 来定义,实验数据的积累使得相应预测算法的开发成为可能 。近年来,在 ΔΔG 预测方面已付出了巨大努力 。目前的方法主要可分为机理预测器、机器学习预测器和深度学习预测器 。然而,在一个新发布的基准数据集S669上进行评估时,这些方法并未展现出相较于传统方法的绝对优势 。关键问题在于训练数据量有限(约10,000条),这对于拥有数百万参数的深度学习模型来说,很容易导致过拟合 。此外,可用的实验数据偏向于少数几个热点蛋白家族、特定的目标氨基酸突变和/或去稳定突变 。这种偏见的一个体现是,目前大多数预测器无法再现 ΔΔG 的反对称性(anti-symmetry)特性,即交换突变体和野生型蛋白状态时,其值应反转 。相较于 ΔΔG 预测,ΔT_m 预测的研究相对较少 。现有模型,如AUTO-MUTE和HoTMuSiC,使用物理化学性质或统计势作为特征,并结合基于机器学习的方法进行预测 。据我们所知,该领域尚无基于深度学习的方法,这可能是因为与 ΔΔG 数据相比,可用的 ΔT_m 数据量大幅减少 。除了面临与 ΔΔG 预测相同的挑战外,ΔT_m 预测的另一个困境是缺乏已建立的开源基准数据集,这阻碍了模型性能的公平比较 。
在本文中,我们首先描述了一个基于几何学习的模型GeoFitness的开发,用于预测蛋白质适应性 。具体来说,我们设计了一种特殊的损失函数,允许使用DMS数据库中的多标签适应性数据来训练一个统一的模型 。由此得到的模型避免了在实际使用前需要重新训练的限制,同时实现了比其他顶尖方法(如ECNet)更优的性能 。此外,通过复用GeoFitness的几何编码器,我们开发了两个额外的下游模型,GeoDDG和GeoDTm,分别用于预测蛋白质突变后的 ΔΔG 和 ΔT_m 。
我们通过两种策略解决了 ΔΔG 和 ΔT_m 预测中数据有限的挑战:一是通过数据收集来扩展训练数据,二是通过继承在DMS数据库上预训练的GeoFitness模型的几何编码器 。特别是后一种策略,考虑到蛋白质变体的适应性数据在数量上比 ΔΔG 和 ΔT_m 的数据总和还要多至少一个数量级,以及蛋白质适应性在生物学上与稳定性的相关性,极大地提升了模型性能和泛化能力 。在基准测试集上(ΔΔG 使用S669,ΔT_m 使用本文自建的S571)进行评估时,GeoDDG和GeoDTm在预测值与实验值的斯皮尔曼相关系数方面,分别比其他顶尖方法高出至少30%和70% 。
2 Results
Overview of the prediction models
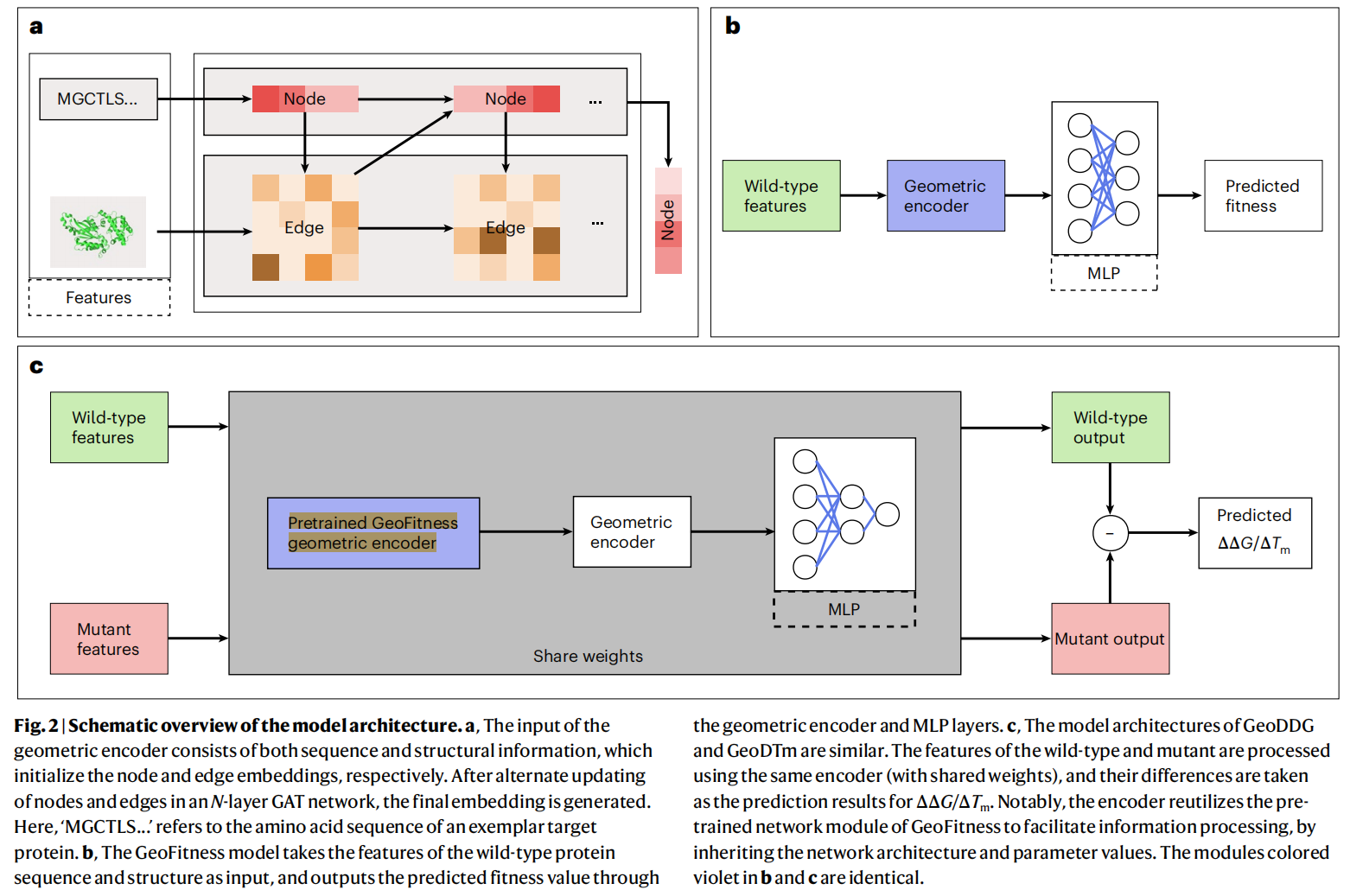
GeoStab-suite 套件包含三个独立的程序——GeoFitness、GeoDDG 和 GeoDTm 。所有这些程序都将来自蛋白质序列和结构的信息聚合到一个基于几何学习的编码器中用于预测 。该几何编码器采用图注意力(GAT)神经网络架构,其中节点(一维)代表氨基酸残基,边(二维)反映残基间的相互作用 (如图 2a 所示 )。
- GeoFitness 是一个统一的模型,能够预测蛋白质所有单一突变体的适应性景观 (如图 2b 所示 )。
- GeoDDG 和 GeoDTm 则复用了 GeoFitness 的预训练信息提取器,来分别预测蛋白质在任意数量突变下的
ΔΔG和ΔT_m值 (如图 2c 所示 )。
蛋白质结构信息既可以来源于蛋白质数据银行(PDB)的实验结构,也可以仅基于序列使用 AlphaFold2 进行预测 。因此,我们训练了 GeoDDG 和 GeoDTm 的两个版本,分别以后缀 ‘-3D’ 和 ‘-Seq’ 来标注:‘-3D’ 版本依赖实验结构,而 ‘-Seq’ 版本在实际使用中仅需序列信息 。
Evaluation of protein fitness prediction by GeoFitness
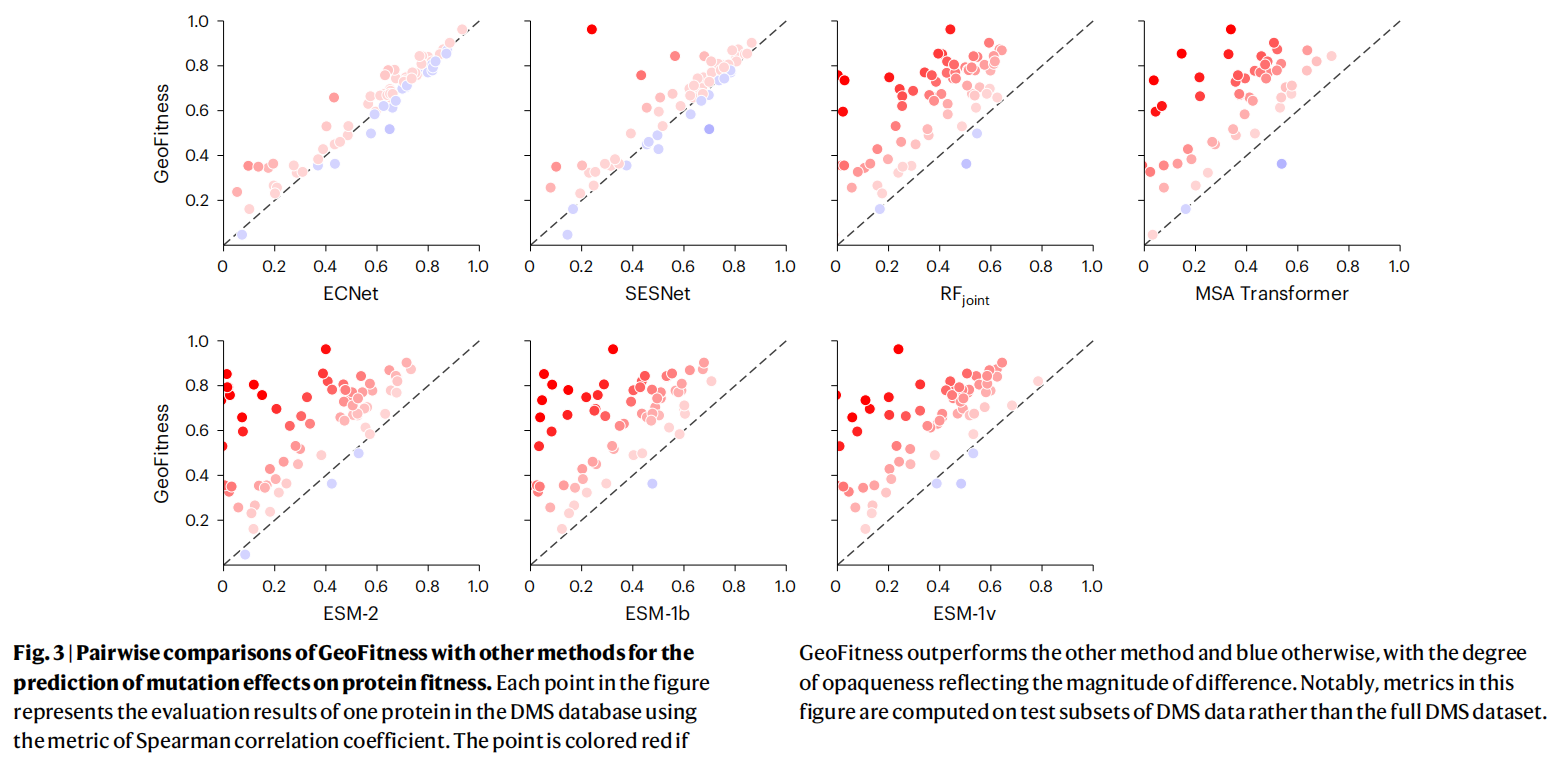
为了便于模型训练,我们将多标签的适应性数据统一转换为每个蛋白质在每个独立位置上所有可用突变的相对排序 。因此,最终的 GeoFitness 是一个通用的适应性预测模型,在实际预测前无需使用目标蛋白的额外适应性数据进行重训练,即允许零样本预测(zero-shot prediction) 。
- 与无监督模型的比较:我们首先将 GeoFitness 与其他完全不依赖适应性数据的统一模型(如 RFjoint、MSA Transformer、ESM-1b、ESM-1v 和 ESM-2)进行了比较 。如图 3 所示,GeoFitness 明显优于这些无监督模型,对 DMS 数据库中几乎所有蛋白质都实现了更高的斯皮尔曼相关系数,这表明在模型训练中使用适应性数据显著提升了模型性能 。
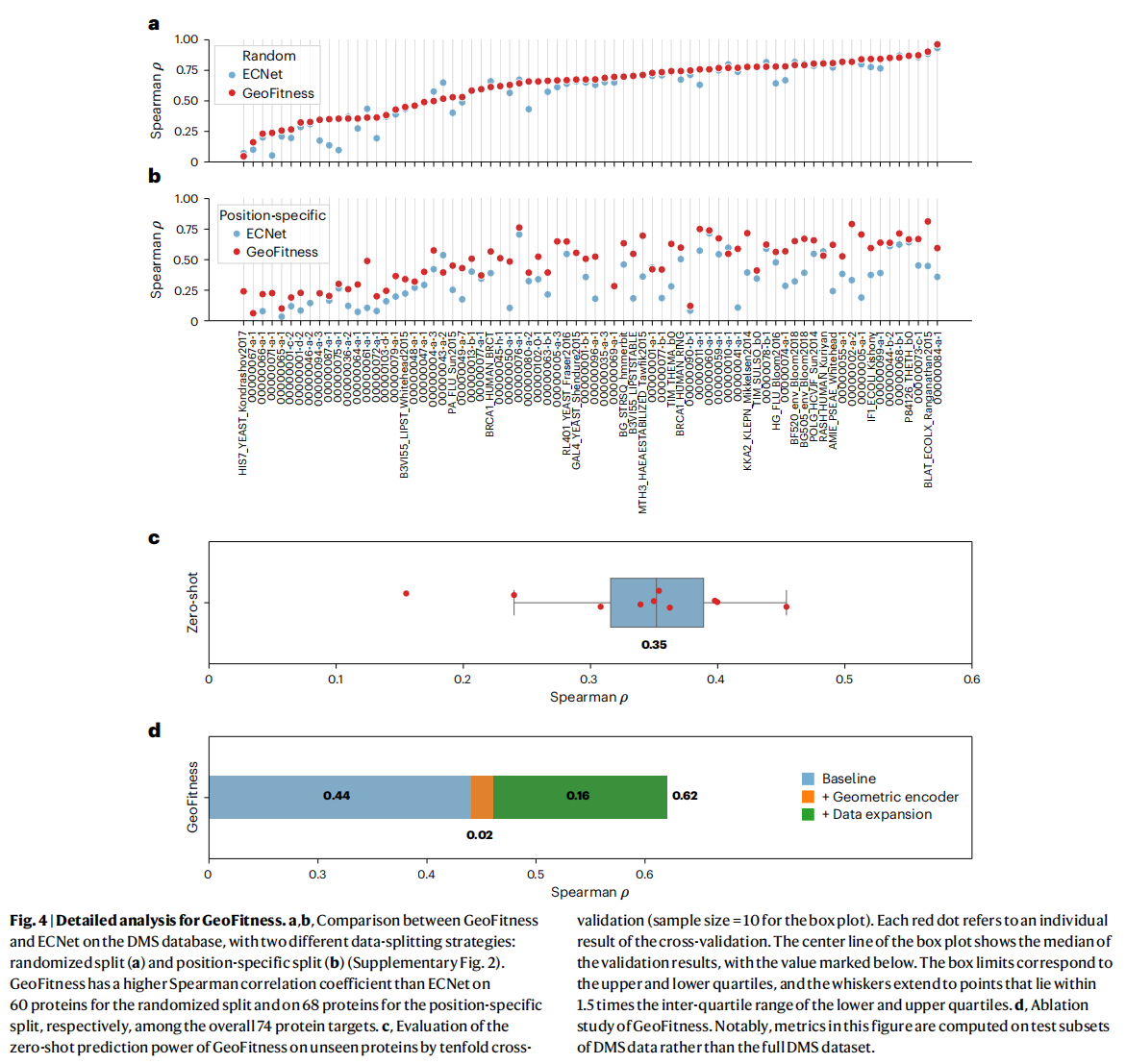
- 与监督模型的比较:随后,我们将其与 ECNet 和 SESNet 进行了评估,这两种顶尖方法需要为每个独立蛋白质训练一个特定模型 。尽管这种比较对我们的方法不公平,GeoFitness 仍在超过一半的测试蛋白质上表现出更好的性能 (如图 3 和 4a 所示 )。在整个DMS数据库上取平均值,通用的 GeoFitness 模型实现的斯皮尔曼相关系数(0.62)略高于一系列经过专门优化的 ECNet(0.59)和 SESNet(0.57)模型 。
- 模型泛化能力:当从默认的随机数据划分策略切换到更能考验模型学习难度的“按位置划分”策略时,GeoFitness 相对于 ECNet 的优势也相应增加 。我们还通过十折交叉验证评估了其对未见过蛋白质的零样本预测能力,结果显示,即使在这种严苛的场景下,GeoFitness 仍保持了可接受的预测能力(斯皮尔曼相关系数中位数为0.35),进一步证明了其良好的泛化性和广泛的适用性 。
- 消融研究 (Ablation study):对 GeoFitness 的消融研究分析了模型架构和数据扩充的贡献 。结果显示,数据扩充(从 MaveDB 数据库中丰富数据)做出了更大的贡献,使平均斯皮尔曼相关系数增加了0.16 。
Evaluation of ΔΔG prediction by GeoDDG
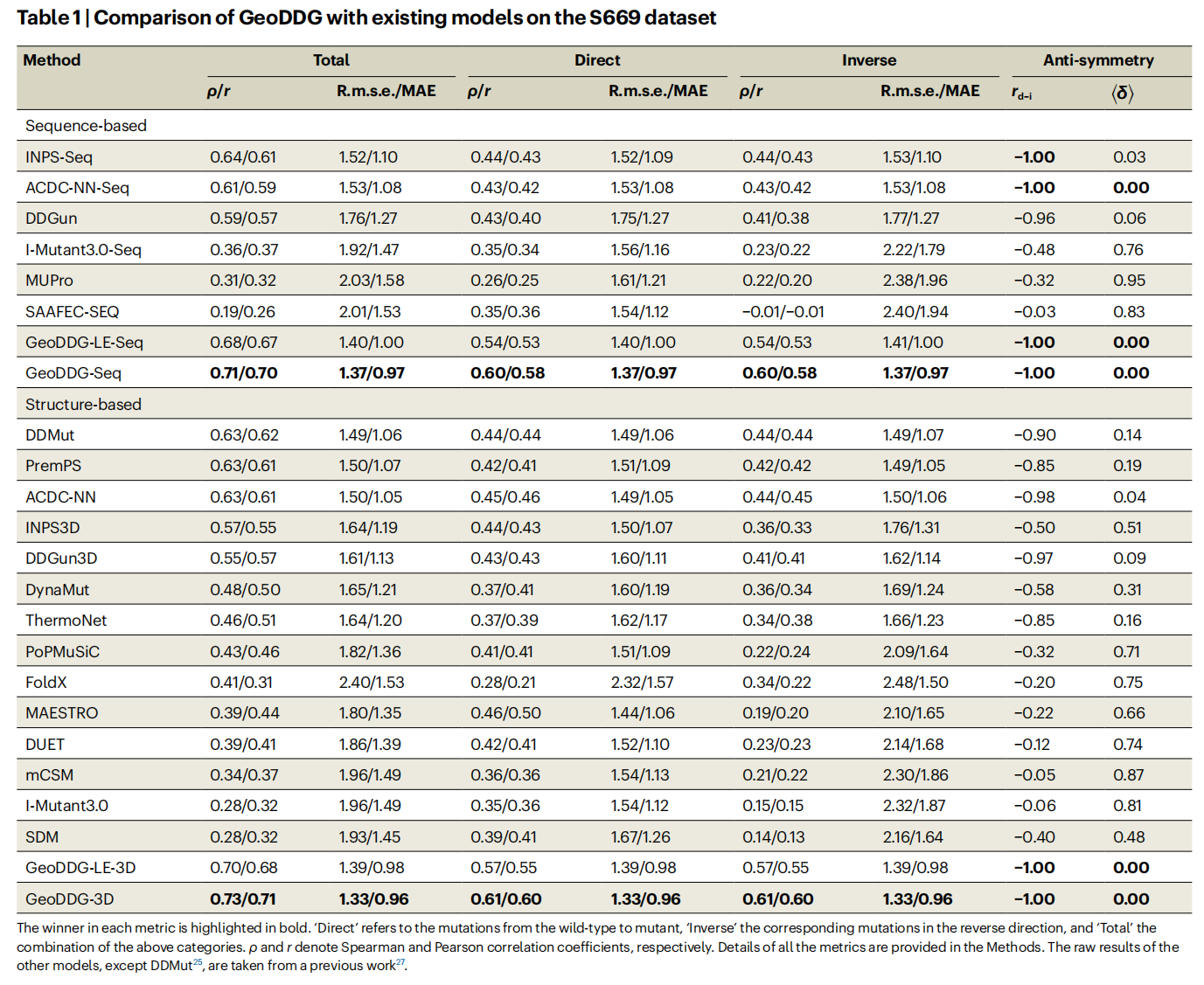
GeoDDG 使用一个共享权重的模块来处理野生型和突变体蛋白的序列与结构信息,并取其差异作为预测结果 。这种设计能够自然地保证预测结果的反对称性 。
- 在 S669 基准上的表现:我们在 S669 基准测试集上对 GeoDDG-Seq 和 GeoDDG-3D 的性能进行了评估,并严格移除了训练集与测试集之间的冗余,以保证评估的公平性 。如表1所示,GeoDDG-Seq 和 GeoDDG-3D 在各自类别(基于序列/基于结构)的所有指标上均优于其他方法 。特别是在斯皮尔曼相关系数上,它们相较于第二名的方法分别取得了 36.4% (0.60 vs 0.44) 和 32.6% (0.61 vs 0.46) 的相对提升 。此外,GeoDDG 是本次测试中唯一能将平均绝对误差(MAE)降至 1 kcal mol⁻¹ 以下的方法 。
- 在有限数据上的表现:为了验证模型的创新点,我们还在一个较小的数据集 S2648 上重新训练了 GeoDDG,命名为 GeoDDG-LE 。结果显示,其性能虽略有下降,但仍然大幅领先所有其他方法,表明我们的方法即使在训练数据有限的情况下也能生成可靠的预测模型 。
- 消融研究:对 GeoDDG 的消融研究(见扩展数据图1和2)表明,在所有改进中,预训练策略(pre-training strategy)带来了最大的性能提升(在GeoDDG-Seq中提升了0.07,在GeoDDG-3D中提升了0.06)。这有力地证明了使用大量蛋白质适应性数据进行模型预训练,可以有效克服过去多年来困扰
ΔΔG预测的数据有限挑战 。
Evaluation of ΔT_m prediction by GeoDTm
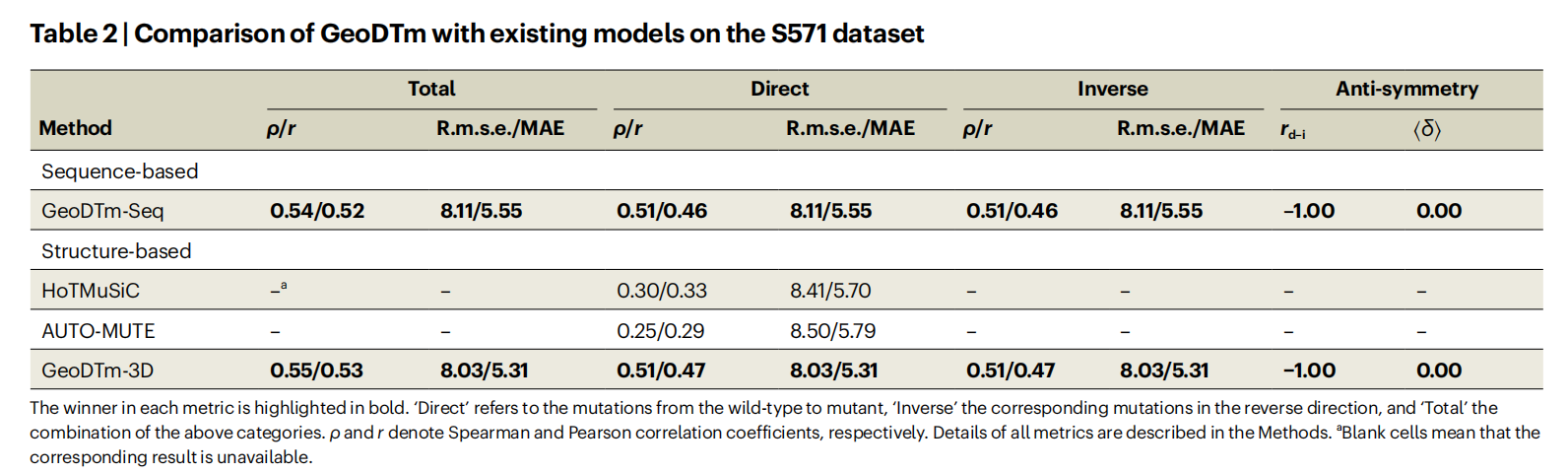
与 ΔΔG 预测不同,ΔT_m 预测领域此前没有已建立的基准测试集 。为解决此问题,我们整理并创建了一个名为 S571 的测试集,它与当前可用的训练数据集无冗余,从而可以进行公平评估 。
- 在 S571 基准上的表现:我们在 S571 测试集上对 GeoDTm-Seq 和 GeoDTm-3D 进行了评估,并与两个现有的
ΔT_m预测器 AUTO-MUTE 和 HoTMUSIC 进行了比较 。如表2所示,GeoDTm-3D 在所有指标上均优于其他基于结构的方法 。值得注意的是,我们的方法将斯皮尔曼相关系数从0.30大幅提升至0.51,相对提升超过70% 。 - 消融研究:对 GeoDTm 的消融研究(见扩展数据图3)再次证实了预训练策略的重要性 。在 GeoDTm 的两个版本中,预训练策略都做出了最大的贡献,将斯皮尔曼相关系数提升了0.09 。这一提升进一步证明了使用大量相关数据进行预训练在消除过拟合和改善模型泛化能力方面的作用 。
Discussion
理解突变效应是蛋白质设计与工程实践中的一个重要问题,因此,开发能够准确高效地学习和预测这些效应的计算方法备受关注 。尽管在模型架构设计上持续努力,但不充分和低效的数据利用已逐渐成为阻碍模型性能进一步提升的瓶颈 。在适应性预测领域,尽管大量的DMS数据为深度学习模型的训练铺平了道路,但由于对多标签数据处理不当,现有模型不得不在预测能力或模型易用性之间做出牺牲 。在 ΔΔG 和 ΔT_m 预测领域,虽然问题定义清晰且数据是单标签的,但数据的有限规模和偏见阻碍了深度学习模型在不发生过拟合的情况下进行训练 。一篇系统性综述甚至声称,根据在 S669 测试集上的评估结果,ΔΔG 预测模型的平均准确性在过去15年中并未得到改善 。对于 ΔT_m 预测,情况甚至更糟,因为客观的测试集(如 ΔΔG 预测中的 S669)付之阙如,这使得对 ΔT_m 预测方法进行公平评估成为不可能 。
在本文中,我们采用了一种可微分的排序算法来构建一个软斯皮尔曼相关系数损失函数,这使得我们能够用所有的DMS数据来训练一个统一的神经网络模型,而不受其多标签性质的影响 。这种损失函数使我们能够充分利用DMS数据集对GeoFitness进行监督式训练,使其能够以高于现有顶尖模型的准确性预测目标蛋白的适应性图谱,且无需在实际使用前用大量同类数据进行严格的模型重训练 。由于蛋白质的稳定性在生物学上与蛋白质的适应性密切相关,我们利用了在适应性数据上预训练好的GeoFitness几何编码器来指导 ΔΔG 和 ΔT_m 的预测 。消融研究表明,该预训练策略分别为 ΔΔG 预测和 ΔT_m 预测的斯皮尔曼相关系数带来了0.07/0.06和0.09的提升,这表明其在缓解有限数据带来的负面影响和提高模型泛化能力方面发挥了显著作用 。
在现阶段,我们仅使用了DMS数据库中的单点突变数据来训练GeoFitness 。显然,如果未来能够纳入多点突变数据,并明确考虑突变位点之间的上位效应(epistatic effects),有望为GeoFitness模型的学习能力带来额外的提升 。这将使其能够更准确地捕捉普适的突变效应,从而进一步惠及下游的蛋白质稳定性预测,或其他因训练数据更稀少而学习难度更大的任务,例如预测蛋白质变体的发光强度和表达水平 。
为了进一步提高蛋白质稳定性的预测能力,我们从ProThermDB和ThermoMutDB中收集、清洗并手动核对了数据,整理出了大型的开源蛋白质稳定性数据集:用于 ΔΔG 的 S8754 和用于 ΔT_m 的 S4346 。对GeoDDG和GeoDTm的消融研究也证实了数据扩充对模型性能的积极影响 。此外,我们构建了一个基准测试集 (S571),以便在 ΔT_m 预测方面进行公平的模型评估 。这三个数据集,S8754、S4346和S571,均可免费下载,并可能惠及整个蛋白质稳定性预测领域 。
本文提出的模型,GeoFitness、GeoDDG和GeoDTm,都基于几何编码器,这是一种基于几何学习的网络架构,旨在同时处理蛋白质序列和结构的信息,并在与下游网络集成时能够实现反对称性预测 。根据对现有模型的评估(表1和表2),利用结构信息明显有益于稳定性预测(通常基于结构的模型性能优于基于序列的模型),但这引入了额外的限制,因为目标蛋白的实验结构往往是不可用的 。因此,我们训练了两个版本的蛋白质稳定性预测器,GeoDDG/GeoDTm-3D和GeoDDG/GeoDTm-Seq,它们分别从实验结构和AlphaFold2预测的结构中获取结构信息 。在实际预测中,当有可靠的实验结构可用时,建议使用 ‘-3D’ 版本以获得更好的性能,而当只有序列信息已知时,‘-Seq’ 版本则是一个选择 。
值得注意的是,在本文提出的稳定性预测器中,我们的 ‘-Seq’ 版本的预测能力非常接近 ‘-3D’ 版本(表1和表2),这表明将来自AlphaFold2的精确预测结构作为空间约束(例如,本文中的边嵌入)进行适当利用,可以有效地辅助如稳定性预测等下游任务,这与之前的发现相符 。
我们已经为GeoStab-suite(包含GeoFitness、GeoDDG和GeoDTm三个预测器)建立了一个网络服务器 。我们期望GeoStab-suite能成为蛋白质科学领域研究人员的一个有用工具 。
Methods
Protein fitness database and dataset
- MaveDB: MaveDB是一个用于存储大规模突变效应数据的开源数据库 。我们从中选择了52个关于单点突变效应的DMS研究 。
- DeepSequence 数据集: 我们也从DeepSequence数据集中筛选了DMS数据,收集了单点突变数据,并排除了与MaveDB重复的部分 。通过这种方式,我们获得了22个DMS数据集 。
- 训练、验证和测试集的划分: 本文使用的DMS数据总共包含来自74个蛋白质的约300,000个条目 。对于每个蛋白质,所有可用的DMS数据被随机地以7:1:2的比例分配为训练集、验证集和测试集 。所有蛋白质的训练数据被进一步合并,作为GeoFitness的综合训练集 。值得注意的是,这里使用的DMS数据集在标签上与蛋白质稳定性数据集没有任何重叠,特别是与
ΔΔG/ΔT_m的基准测试集没有重叠 。因此,使用大量的DMS数据进行模型预训练不会给下游的ΔΔG和ΔT_m预测任务或性能评估带来信息泄露 。
Protein stability database and dataset
- ProThermDB: 这是一个蛋白质突变体的热力学数据库,是其前身ProTherm的升级版,包含了约31,500条蛋白质稳定性数据 。
- ThermoMutDB: 这是一个手动整理的蛋白质突变体热力学数据库,包含了约15,000条蛋白质稳定性数据 。
- S2648: 这是一个
ΔΔG数据集,包含从ProTherm数据库收集的131个蛋白质的2,648个单点突变 。它在以往的ΔΔG预测算法中被广泛用作训练集 。 - S669: 这是一个由94个蛋白质的669个单点突变组成的
ΔΔG数据集,与S2648中的蛋白质序列相似性低于25% 。它通常被认为是用于公平评估ΔΔG预测方法性能的基准测试集 。 - S461: 该数据集被认为是S669的一个更干净的子集,保留了S669中461个单点突变并修正了标签,也被用作一个辅助基准测试集 。
- S8754: 这是本文构建的用于
ΔΔG数据预测的训练集,包含301个蛋白质的8,754个单点突变 。我们从ProThermDB和ThermoMutDB收集数据,经过谨慎清洗、合并和手动验证,以保证每个条目的唯一性和高可信度 。在移除了与S669测试集冗余的数据后,我们最终构建了目前用于模型训练的最大ΔΔG数据集 。 - M1261: 这是本文为评估模型在多点突变
ΔΔG值预测中的性能而生成的数据集,由133个蛋白质的792个双突变和469个三突变或更高阶突变组成 。 - S571: 这是本文构建的用于评估
ΔT_m预测方法的基准测试集,包含37个蛋白质的571个单点突变 。我们采用与S8754类似的方法进行数据清洗,并消除了与常用训练集S1626的冗余,从而确保了评估的公平性 。 - S4346: 这是本文构建的用于
ΔT_m预测的训练集,包含349个蛋白质的4,346个单点突变 。在移除了与S571测试集的冗余后,我们构建了目前用于模型训练的最大ΔT_m数据集 。
Information processing in the geometric learning-based model architecture
如图2a所示,几何编码器采用GAT架构,使用节点和边分别处理来自蛋白质序列和结构的输入特征 。
- 序列特征: 来源于大规模蛋白质语言模型ESM-2 。GAT中的每个节点主要由ESM-2输出的相应残基的嵌入(embedding)来初始化 。
- 结构特征: 每条边由蛋白质3D结构中一对残基之间的相对几何关系来初始化 。
- 信息融合与更新: 在几何编码器的每一层中,节点嵌入和边嵌入通过一系列运算(包括多头注意力机制)进行融合和更新 。
- 反对称性保证: 在GeoDDG和GeoDTm的模型架构中(图2c),野生型特征和突变体特征由权重共享的同一个神经网络处理 。因此,当输入特征交换时,野生型和突变体蛋白输出值之间的差异理应改变符号,这种设计自然地保证了预测结果的排列反对称性 。
Training details
- 数据集使用: 我们使用DMS数据库训练、验证和测试GeoFitness 。使用S8754作为GeoDDG的训练集,S669作为测试集 。使用S4346作为GeoDTm的训练集,S571作为测试集 。训练集和测试集之间的冗余已通过严格标准(训练集中的蛋白质与测试集中的蛋白质序列一致性>25%的均被移除)仔细清除 。
- 结构来源: 我们训练了两个版本的GeoDDG/GeoDTm:使用实验野生型结构的(GeoDDG-3D/GeoDTm-3D)和使用AlphaFold2预测的野生型结构的(GeoDDG-Seq/GeoDTm-Seq) 。
- 特征生成: 我们选择了ESM-2的33层650M参数模型来生成每个残基的“词”嵌入 。利用FoldX软件基于野生型结构预测突变体的结构 。
- 损失函数:
- 在GeoFitness的优化中,为了利用多标签DMS数据,我们将所有适应性数据转换为每个独立位置上所有可能突变的相对排名 。我们采用可微分的软斯皮尔曼相关系数作为损失函数,这使得在神经网络中进行梯度更新成为可能 。
- 在GeoDDG和GeoDTm的模型训练中,我们采用了一个联合损失函数,它同时包含了软斯皮尔曼相关系数和预测值与实验值之间的均方误差(m.s.e.) 。
- 微调过程: 在训练GeoDDG和GeoDTm时,继承自GeoFitness的预训练几何编码器的参数在初始阶段被冻结,以便快速优化其他参数 。随后,在微调阶段,所有参数都被允许进行微小调整 。