论文地址:evedesign accessible biosequence design with a unified framework
evedesign:统一框架生物序列设计
Abstract
蛋白质工程的机器学习方法很少具有互操作性,通常需要定制的工作流,并且对非专业人员来说仍然难以使用 。然而,当前最重要的一些设计问题——如受限于现实世界约束的条件设计、多目标优化,以及通过实验数据不断改进后续设计轮次的迭代式“实验室在环(lab-in-the-loop)”工作流——恰恰需要任何单一工具都无法提供的那种灵活且可组合的基础设施 。为此,提出了 evedesign,这是一个统一的开源框架,它以一种与具体方法无关的方式将条件生物序列设计形式化,从而能够根据标准化规范将监督模型和无监督模型结合起来,实现复杂的多目标工作流,并且该框架从构建之初就旨在支持迭代式的实验整合 。此外,一个交互式的 Web 界面(https://evedesign.bio)为广泛的科学受众促进了端到端的设计流程 。在抗体工程、酶设计以及天然酶发现中展示了 evedesign 的实用性,并邀请开源社区共同参与贡献
1 Introduction
作者首先指出,蛋白质工程具有解决下一代疗法、可持续生物制造、生物安全、污染物修复和气候适应等关键未满足需求的巨大潜力 。近年来,用于蛋白质设计的机器学习方法呈现出爆发式增长,涵盖了在进化序列上训练的基于 MSA(多序列比对)的方法、在未对齐蛋白质序列上训练的大语言模型(LLM)、逆折叠模型以及 de novo 3D 结构设计模型等 。尽管这些方法在实验中取得了令人瞩目的成果,但即使对于专家而言,将这些方法转化为实际的设计工作流依然十分困难,并且持续需要定制化、高劳动强度的代码开发 。
研究团队提出,有两个相互关联的缺口限制了这一领域进展的实际应用价值 :
- 第一个缺口是缺乏标准化的编程接口,这意味着各种方法无法轻易地进行比较、组合或替换 。这种互操作性的缺乏在条件设计(Conditional design)中代价尤为高昂——在实际场景中,蛋白质设计需要满足诸如热稳定性、pH 耐受性、清除 T 细胞表位或与新靶点结合等现实世界约束,并且需要能够将新实验数据纳入其中以指导后续设计轮次的迭代式“实验室在环(lab-in-the-loop)”工作流 。这些多目标、约束驱动的任务是大多数现实世界设计项目的核心,然而现有的框架要么不是与模型无关的,要么无法推广到单体蛋白质基于序列的模型之外 。此外,这种缺乏互操作性的问题还延伸到了将模型受控集成到智能体系统(agentic systems)中 。
- 第二个缺口在于目前缺乏一个开源的、交互式的用户界面,能够跨越从目标蛋白质到可订购 DNA 序列的完整设计工作流,同时保持独立于底层模型和流水线的适用性 。虽然市场上存在一些专有(商业)解决方案,但由于缺乏开源的替代方案,迫使非计算领域的研究人员只能依赖相互孤立的工具,这限制了整个科学社区的复现性与可及性 。
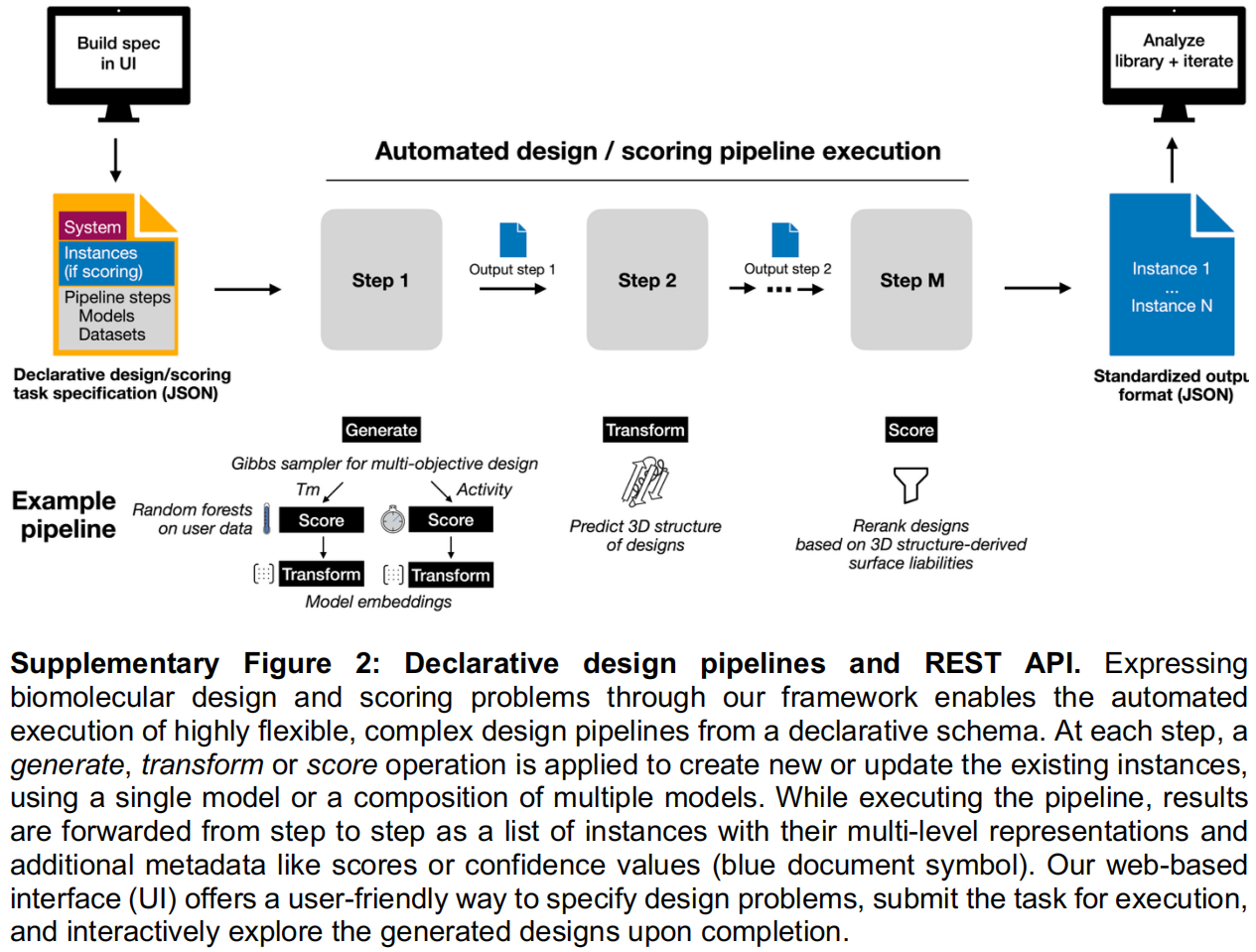
为了填补这些缺口并使蛋白质设计符合 FAIR 引导原则(可发现、可访问、可互操作、可重用),作者推出了 evedesign 。这是一个多层的开放式框架,主要由以下三个部分组成 :
- 一个标准化的模型接口,并在 Python 包中提供了参考实现;
- 一个 REST API 和流水线运行器,用于通过声明式作业规范执行设计任务;
- 一个覆盖完整设计工作流的交互式用户界面(公开访问地址为 https://evedesign.bio )。
最后,作者强调,该框架被设计为一个具有生命力的社区资源:其模块化架构允许随着领域的不断发展而融入新的模型和工作流,从而确保 evedesign 对于计算研究人员和实验研究人员而言,都是一个紧跟前沿且具备高扩展性的解决方案 。
2 Results and Discussion
A unified interface for biomolecular design
研究团队指出,阻碍不同蛋白质设计方法实际应用的核心障碍,是缺乏一种通用的底层语言。为此,evedesign 构建了一套统一接口:
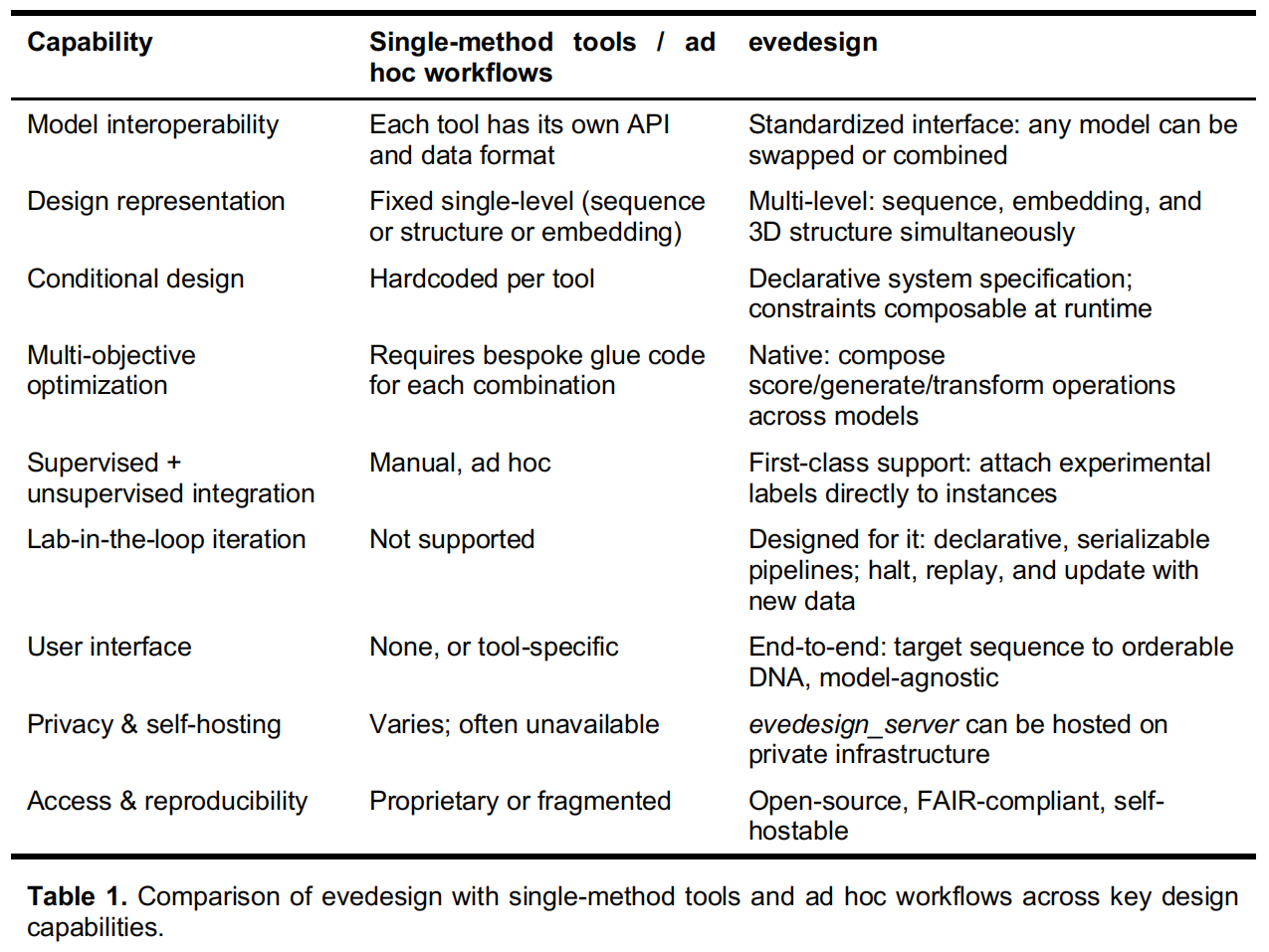
Framing design as a conditional modeling problem)
作者将生物分子设计框架化为一个条件建模问题。在这一框架下,用户无需关心底层模型的具体格式,只需通过一个标准化的、声明式的数据结构,指定由个体实体(如蛋白质、DNA 或 RNA 链、配体)组成的分子系统。 用户可以为每个实体提供任意已知的条件信息,例如一级序列、3D 结构、同源物、结合伴侣、翻译后修饰或多聚体状态。这种设置极具灵活性:序列可以被完全指定、部分掩蔽或完全开放以供设计,结构也可以包含片段或替代构象状态。 在这种范式下,整体的设计目标(例如在消除 T 细胞表位的同时优化热稳定性)不需要通过额外的代码显式声明,而是自然而然地从用户选用的模型以及施加在模型上的系统级约束中呈现出来。
A multi-level instance representation)
当针对特定的条件建模问题设定好之后,模型将作用于具体的“实例(instances)”(即分子系统的具体形态,如某个候选突变体或新生成的序列)。 每个实例都携带一种多层级的表征,能够同时编码三个级别的信息:序列、位置嵌入(Embedding)以及 3D 结构(PDB 格式)。与那些强制使用单一固定表征的传统框架不同,这种层级方案使得设计结果能够在基于不同输入类型的模型之间无缝流动。 例如,同一个工作流水线可以同时使用序列似然性和 3D 结构约束来为设计方案打分,或者将来自大语言模型(LLM)的嵌入映射到坐标上进行结构验证。此外,这种实例格式还天然支持用于“实验室在环”应用的监督学习工作流:实验测量值可以直接作为标签附加到实例上,而无监督模型产生的嵌入或似然值则可作为输入特征,用于训练能够预测目标属性的回归模型。
Three composable operations
为了在不编写定制代码的情况下覆盖各种设计和预测任务,作者提出,所有的工作流都可以通过组合三个标准化的模型操作来表达(其语义类似于通用的机器学习框架 scikit-learn,但专门适配了多层级实例表征):
- Generate(生成):以系统规格为条件约束,在模型偏好的表征层级上产生新的设计实例。
- Score(打分):为每个实例分配一个定量的适应度值(优选对数似然值),从而使得不同设计方案或突变体之间能够相互比较。
- Transform(转换):在不同的表征层级之间映射实例,例如从序列预测 3D 结构,或者通过序列计算嵌入向量。
因为所有的操作都共享同一种标准化的实例格式,一个模型的输出可以直接被另一个模型作为输入消费,且无需重新格式化。这种特性使得研究人员能够通过简单的组合组装出复杂的多目标工作流。
The evedesign software packages
1. 核心 Python 包 (evedesign) 这是整个框架的基础,采用 MIT 开源许可。
- 内置模型实现:它实现了标准化的接口,并包含了三种关键模型类的参考实现:(i) 全新的进化方法 EVmutation2;(ii) 蛋白质语言模型 ESM-2;(iii) 用于逆折叠的 ProteinMPNN/LigandMPNN。
- 丰富的实用工具:除了模型本身,该包还捆绑了一系列通用的辅助工具,包括多实体吉布斯采样器(Gibbs sampler)、序列距离约束、基于 MMseqs2 的 MSA 生成、基于 FoldSeek 的 3D 结构模板搜索与映射、序列空间分析以及密码子优化工具。
- 开源扩展性:研究团队积极邀请开源社区在共享规范下贡献更多附加模型的实现。
2. 服务端与任务调度引擎 (evedesign_server) 在核心包之上,团队开发了同样采用 MIT 许可的 evedesign_server Python 包。
- 架构与功能:它实现了一个 REST API 和一个轻量级的设计流水线执行引擎,用于设计任务的分布式执行和状态跟踪。
- 隐私与私有化部署:对于有商业保密或数据隐私需求的用户,该包允许他们在自己的计算基础设施上托管完整的设计服务器后端,确保敏感的序列和实验数据不会离开用户的本地环境。
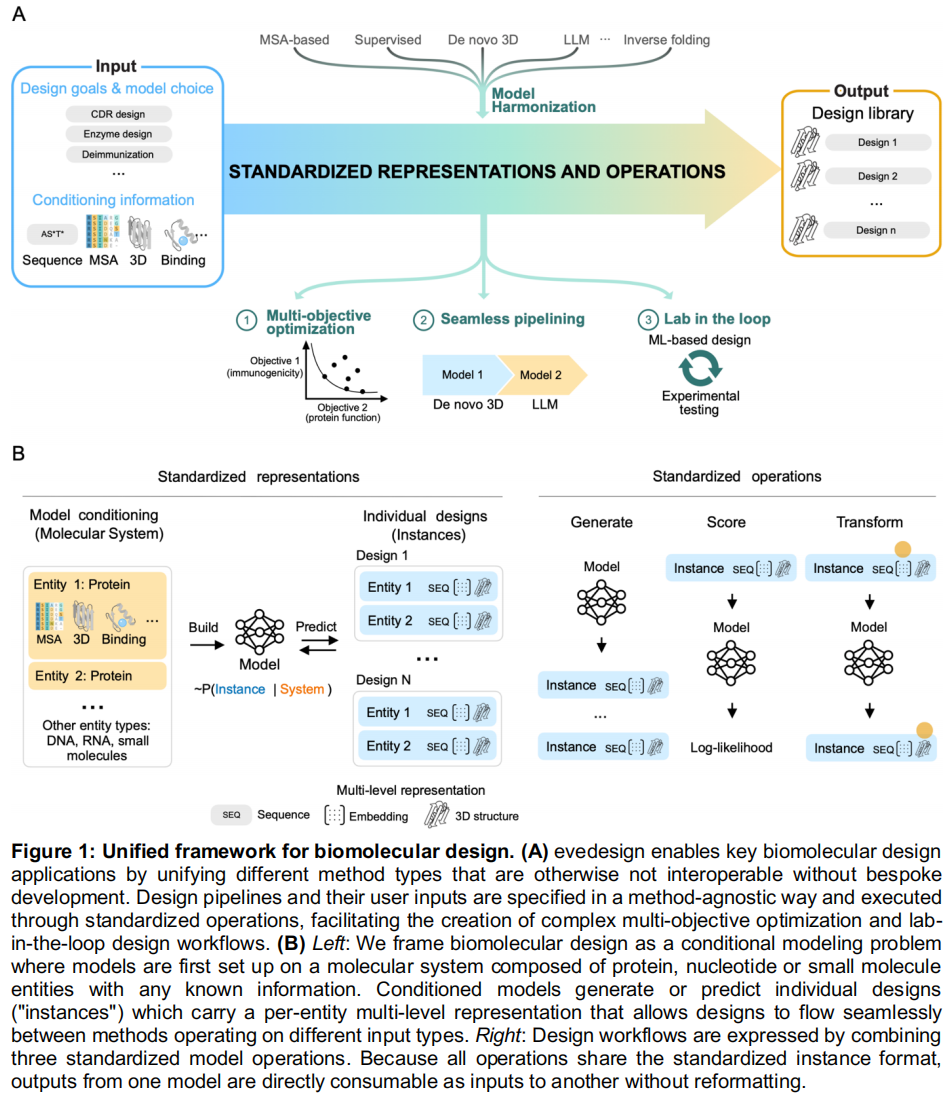
3. 交互式前端应用 (Web UI) 为了让非计算专业背景的研究人员也能轻松使用,团队开发了一个基于 React 的前端应用程序。
- 端到端无代码体验:用户无需编写代码或执行命令行工具,即可通过用户友好的方式完成从目标序列到经过密码子优化的核苷酸序列的全套设计任务。
- 丰富的数据可视化:在任务完成后,用户界面允许在现有 3D 结构和进化序列的背景下,对生成的设计进行交互式探索,并提供丰富的数据可视化功能(如边际氨基酸频率、自然序列空间投影等)。
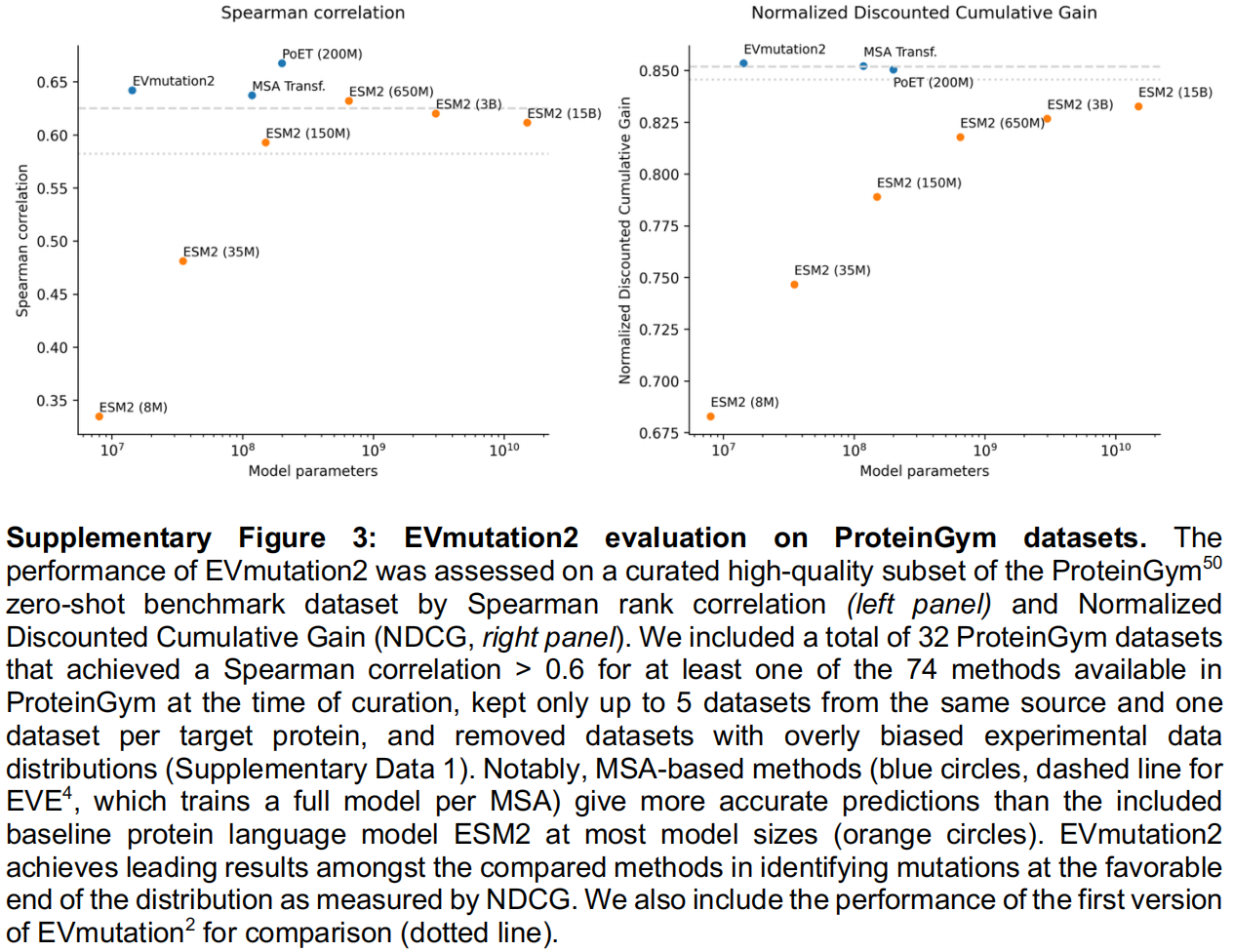
EVmutation2: a lightweight evolutionary model for generative design
虽然进化模型(Evolutionary models)在设计与天然序列差异较大的功能性蛋白质方面有着强大的实验记录,但现有的方法(包括作者之前开发的 EVE 模型)都需要针对单个 MSA(多序列比对)进行专门的训练或微调。这种特性导致它们运行速度太慢,无法直接用于交互式的服务器中。
为了解决这个问题,研究团队专门为快速的生成式推理(fast generative inference)开发了一个全新的基于 MSA 的进化模型——EVmutation2。
以下是该模型的核心技术细节:
- 架构创新:EVmutation2 将一个阶数无关的自回归解码器(order-invariant autoregressive decoder)耦合到了一个紧凑版 AlphaFold3 重新实现的单序列和双序列(single- and pair-sequence)表征上。
- 参数量与注意力机制:该模型非常轻量,仅有 14.3M(1430万)个参数。它超越了 BERT 风格的掩码预测,支持直接的序列采样。解码器的注意力权重不是通过完整的 Query-Key-Value 计算得出的,而是通过对成对表征(pair representation)进行线性投影(linear projection)得出,这既强制模型学习具有生物学意义的成对相互作用,又大幅减少了参数量。
- 上下文最大化:模型会将所有已知的 Token 以随机顺序移动到前缀(prefix)中,从而无论被设计的位置在序列中的哪个地方,都能为其提供最大的上下文信息。
- 训练与性能:该模型在 OpenProteinSet 上进行了训练,通过交叉熵损失来重建随机掩码的 MSA 序列。尽管体积小巧,EVmutation2 在 ProteinGym 基准测试中的表现与 EVE 相当,且不需要针对每个目标进行微调,这使其非常适合作为一个通用的社区服务器模型。
Case studies: demonstrating evedesign workflows
研究团队在案例研究(Case studies)部分明确指出,展示这些案例的目的并非为了宣称模型达到了当前最佳性能(SOTA),而是为了证明 evedesign 的标准化接口、可组合操作以及多层级实例表征能够将各种复杂的真实世界设计任务表达为连贯、可重复的工作流,且完全无需编写定制代码。以下是对这三个案例的重新总结:
Unsupervised enzyme design with an evolutionary model
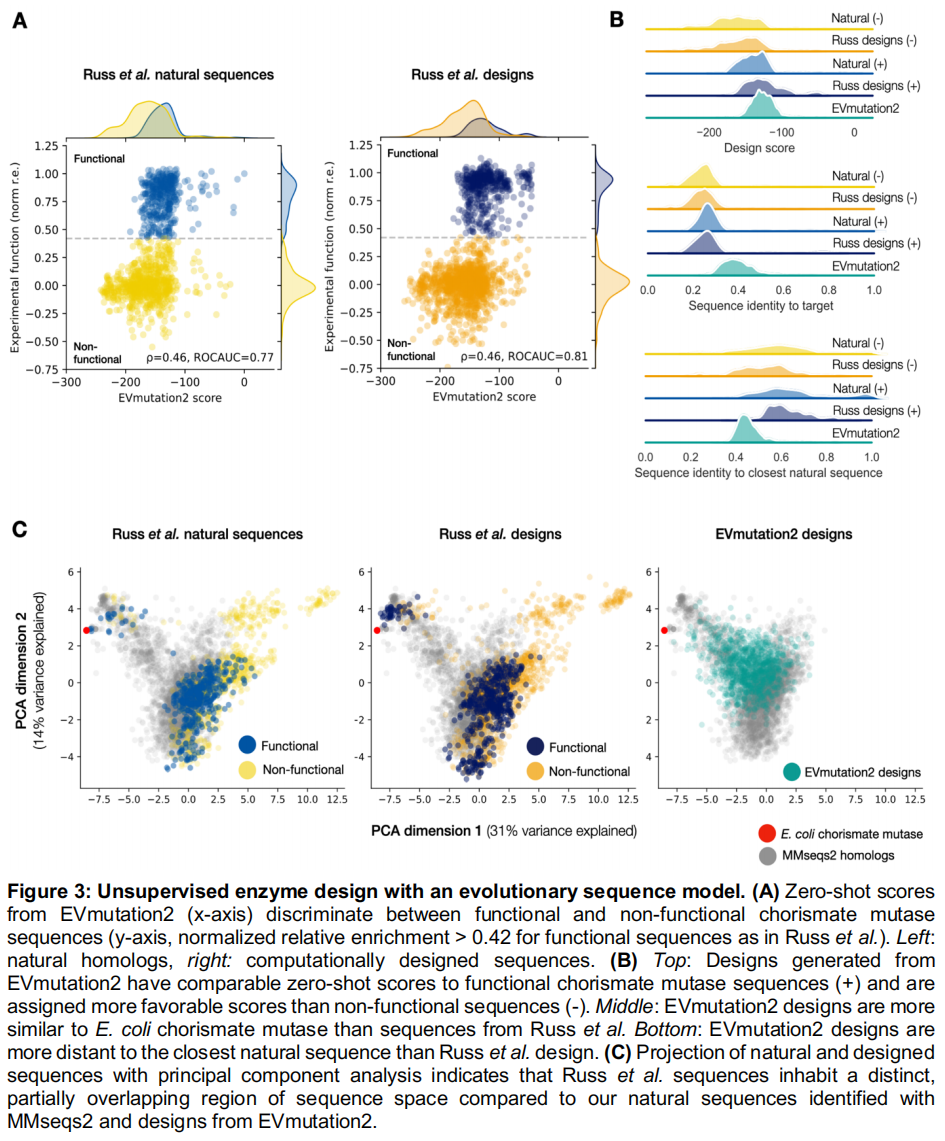
为了演示如何使用前文介绍的进化模型进行生成式序列设计,研究团队复现了一个类似于 Russ 等人所做的工作流。在该研究中,Russ 等人使用 Potts 模型(一种基于统计物理的马尔可夫随机场模型)设计了芳香族氨基酸生物合成关键酶——分支酸变位酶(EcCM)的多样化功能变体,并通过高通量体内互补实验进行了验证。
- 模型构建与验证:研究团队从 MMseqs2 获取的天然 EcCM 同源物多序列比对(MSA)出发,构建了一个 EVmutation2 模型,并证实其零样本(Zero-shot)得分能够在天然同源物集合和 Russ 等人的实验设计中,有效区分功能性与非功能性序列(ROCAUC 分别为 0.77 和 0.81;Figure 3A),这确立了该模型能够捕捉该家族中的功能性约束。
- 生成设计与特征分析:随后,团队利用 evedesign 的生成(
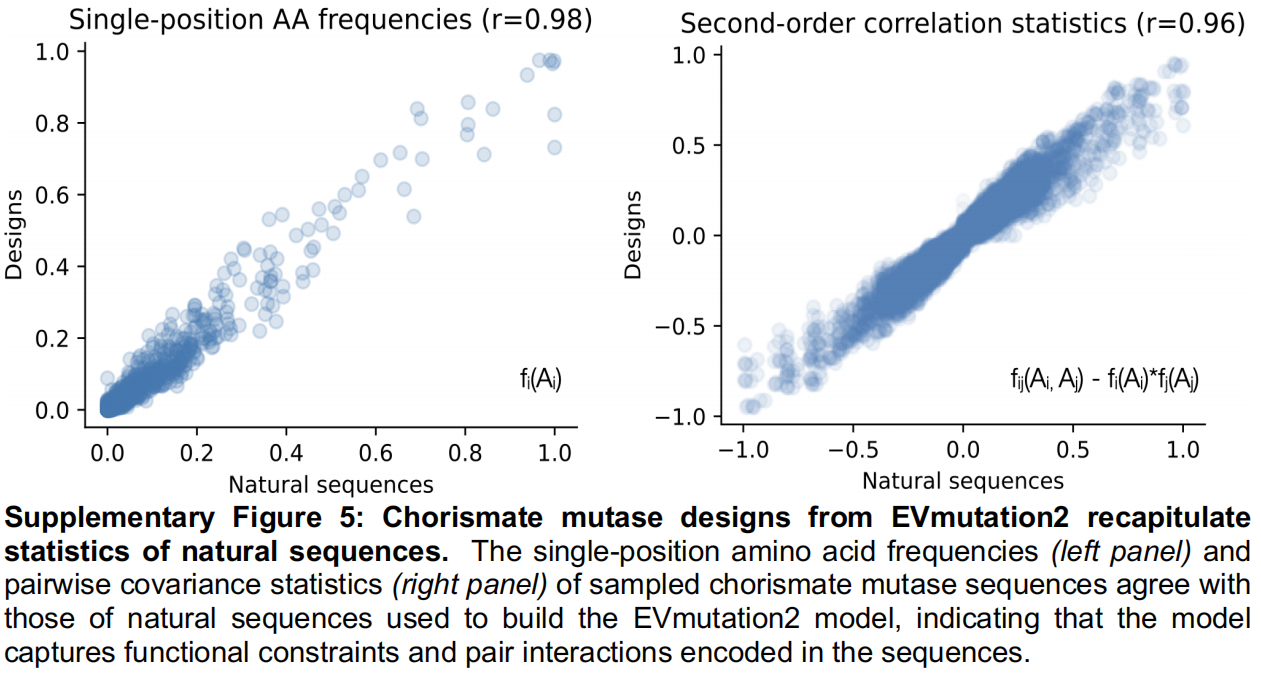
generate)操作,通过自回归采样产生了 1,024 个新序列。这些设计重现了天然序列的一阶和二阶氨基酸统计特性($r=0.98$ 和 $r=0.96$),表明共进化约束得到了保留。其对数似然得分与功能性的天然序列及 Russ 等人的设计相当,且大幅高于非功能性序列(Figure 3B)。在序列空间中,EVmutation2 的设计内插(interpolate)了由 MMseqs2 定义的天然序列分布,且平均而言比 Russ 等人的设计更相似于 EcCM(中位数等同度为 39% 对比 25%),同时依然展现出了对最接近天然序列的有意义的外推(Figure 3C)。 - 结论:该工作流证明了一个完全无监督的生成式流水线如何在 evedesign 中仅用几行代码完成组装和执行。
Complementary sequence- and structure-based antibody scoring
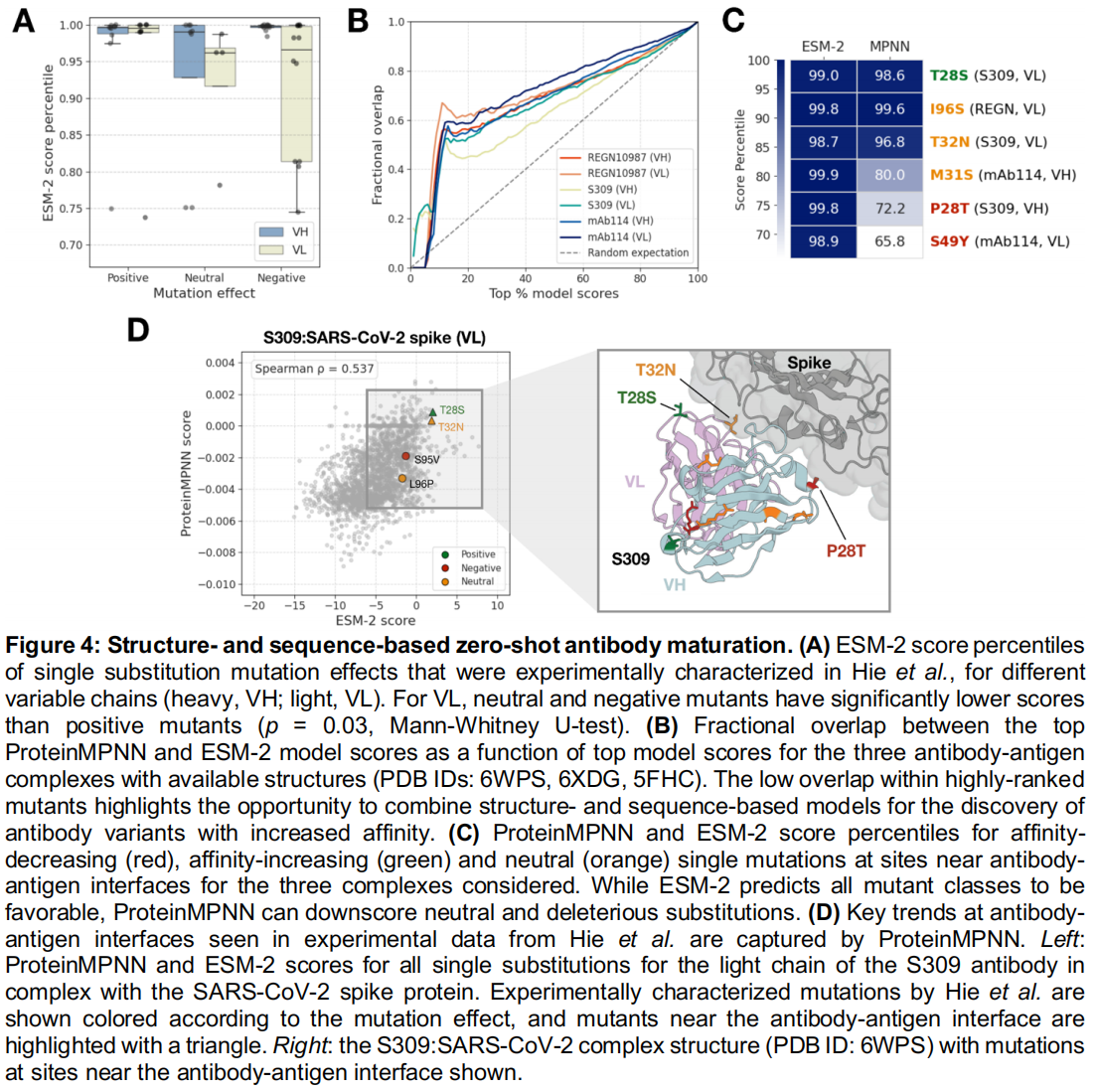
为了说明 evedesign 如何让序列和结构模型在单一工作流中组合,作者对 Hie 等人先前研究的七种临床相关抗体的可变链进行了单残基替换的打分。Hie 等人最初使用了 ESM-1v 集合来进行语言模型引导的亲和力成熟。
- 单纯序列模型的局限:通过应用 evedesign 的打分(
score)操作结合 ESM-2,团队发现 54 个实验测试的有益突变中有 44 个(81.5%)排在了 ESM-2 得分的前 5%(Figure 4A),这与原始发现一致。值得注意的是,ESM-2 为那些降低或不影响抗原结合的轻链突变分配了较低的分数($p=0.03$,Mann-Whitney U-test),尽管这种效应对重链突变并不显著,这是原始 ESM-1 集合未能捕捉到的区别。然而,由于无监督语言模型是在没有结构上下文的情况下针对序列进行训练的,因此不期望它们能够显式地捕捉结合界面的效应。 - 结构模型的互补优势:对于三个具备可用实验测定结构的抗体-抗原复合物,团队额外应用了以 3D 坐标为条件的 ProteinMPNN 的打分操作。ProteinMPNN 和 ESM-2 的得分全局相关,但在最高分突变中表现出极小的重叠(前 5% 的平均重叠度为 0.08;Figure 4B)。重要的是,ProteinMPNN 为界面近端(距离抗原 6Å 以内)的有害和中性突变分配了大幅降低的得分,同时在所有复合物中正确地将唯一的有益界面突变排在高位(Figure 4C, D)。
- 结论:这一分歧说明了多模型 evedesign 框架的一个关键优势:基于序列和基于结构的得分提供了互补的信息,并且可以直接被组合起来对突变进行优先级排序,这对于抗体成熟等对相互作用敏感的应用尤为重要。
Supervised discovery of efficient enzyme variants
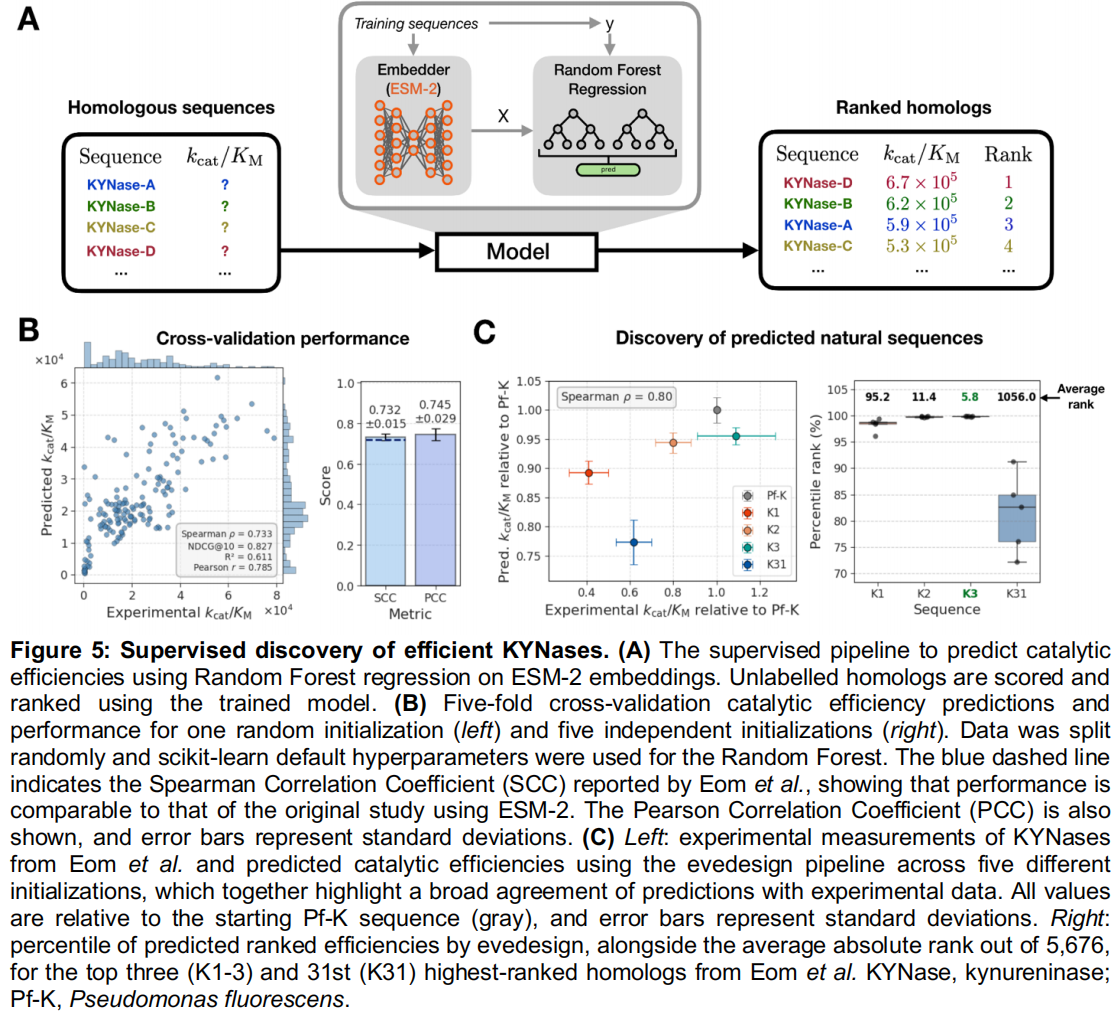
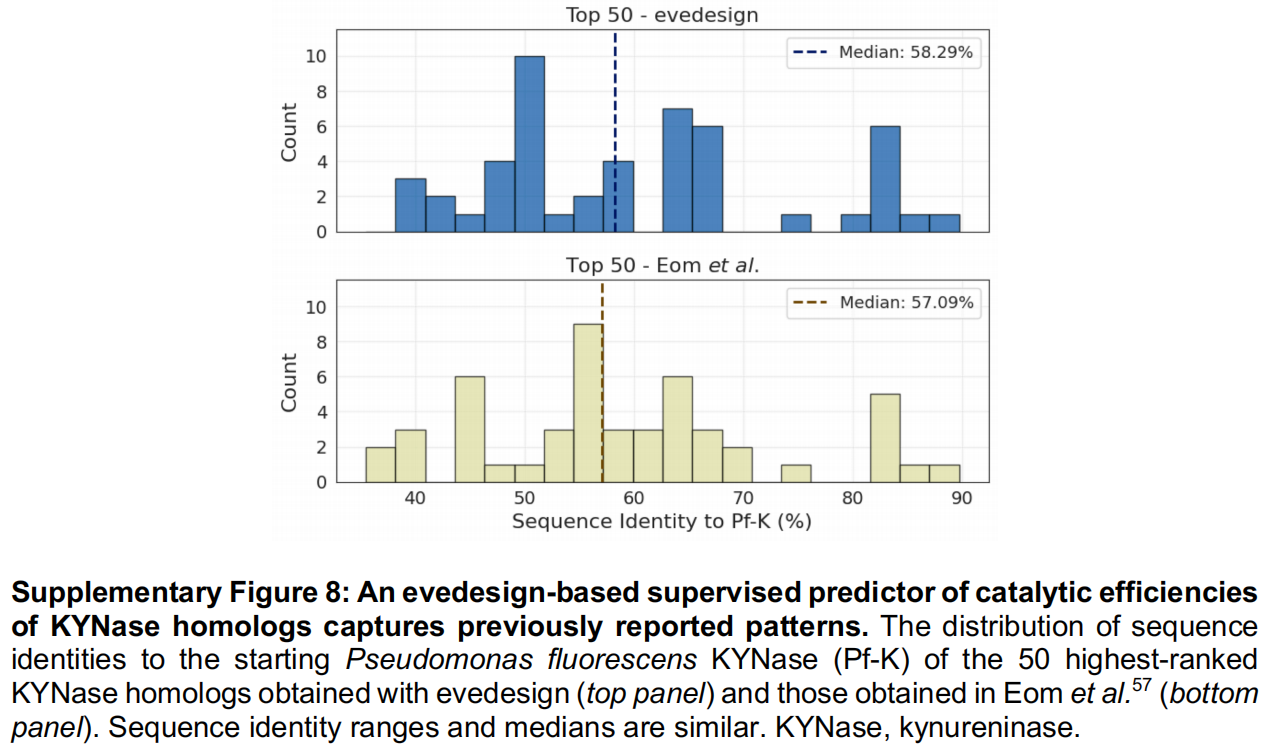
为了演示 evedesign 中的有监督属性预测,作者复现了 Eom 等人的一项分析。Eom 等人在一个经过整理的犬尿氨酸酶(KYNase)动力学数据集上训练了一个基于 ESM-1b 嵌入的随机森林回归器,用于在天然序列数据库中挖掘具有改善催化效率的同源物。
- 模型训练:利用 evedesign 的转换(
transform)操作计算 ESM-2 嵌入,并利用其实例标签方案将实验测定的 $k_{cat}/K_M$ 值作为训练目标附加其上,团队训练了一个等效的回归器,并恢复了与原始研究相匹配的交叉验证性能(Spearman $\rho = 0.73$ 对比 0.72;Figure 5A, B)。 - 数据库规模的挖掘:通过打分(
score)操作对 5,676 个未标记的同源物进行排名,重现了关键的实验结果:最高效的变体(K3)在不同的随机种子下始终排在靠前的前 10 名,而较低效率的变体排名则逐步下降(Figure 5C)。 - 结论:这证实了 evedesign 的有监督工作流能够泛化到数据库规模的属性引导搜索中,而无需对底层框架进行任何修改。
Conclusion
1. 解决碎片化与可访问性壁垒 作者首先重申,evedesign 是一个统一的开源框架,旨在解决目前限制机器学习方法在蛋白质工程中产生现实影响的碎片化和可访问性障碍。通过标准化模型表示分子系统、交换设计以及组合成流水线的方式,evedesign 使得应对治疗开发、生物安全和可持续生物制造中最关键的条件性、多目标设计问题变得切实可行,且无需为每项新任务进行定制化的代码实现。这一点已通过无监督生成设计、互补的基于序列和结构的打分,以及有监督的属性引导发现这三个案例研究得到了验证。
2. 灵活服务于广泛的科研社区 该框架的模块化架构是为服务广泛的社区而有意设计的:
- 计算生物学家和 ML(机器学习)研究人员:可以通过实现少量 Python 接口方法来集成新模型,同时保留对其自身代码的完全控制权。
- 实验学家:可以通过交互式界面 (https://evedesign.bio) 访问上述相同的工作流,降低了使用门槛。
- 有数据隐私要求的用户(如商业或临床环境):
evedesign_server包可以在私有基础设施上进行自托管,确保专有的序列和实验数据永远不会离开用户自己的计算环境。 - 透明度与开源协议:作为一个符合 FAIR 原则、采用 MIT/AGPLv3 许可的平台,evedesign 提供了严格的 ML 引导设计所必需的透明度。
3. 未来愿景与“实验室在环”前沿 最后,作者强调,这里展示的框架是一个具有生命力的软件基础(living software foundation),而不是一个最终的成品。
- 功能扩展:团队计划扩展模型库,包括纳入从头(de novo)结构设计方法。
- 实验室在环(lab-in-the-loop):作者特别指出,流水线运行器的声明式作业规范和可序列化的实例格式,从一开始就是为了支持完全迭代的“实验室在环”工作流而设计的,在这种工作流中,新的实验数据可以不断改进设计循环。作者指出,这是那些临时拼凑的、互不连接的工具集合所无法轻易容纳的能力,并将其完全实现视为该领域最重要、最近期的前沿方向。
- 社区共建:作者积极邀请并欢迎社区做出贡献,以与框架所集成的各种方法一起推动 evedesign 的成长。
Supplementary Figures