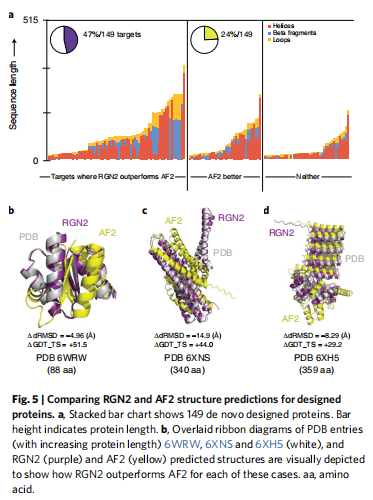
论文地址:Single-sequence protein structure prediction using a language model and deep learning
RGN2:单条蛋白质直接预测结构
Abstract
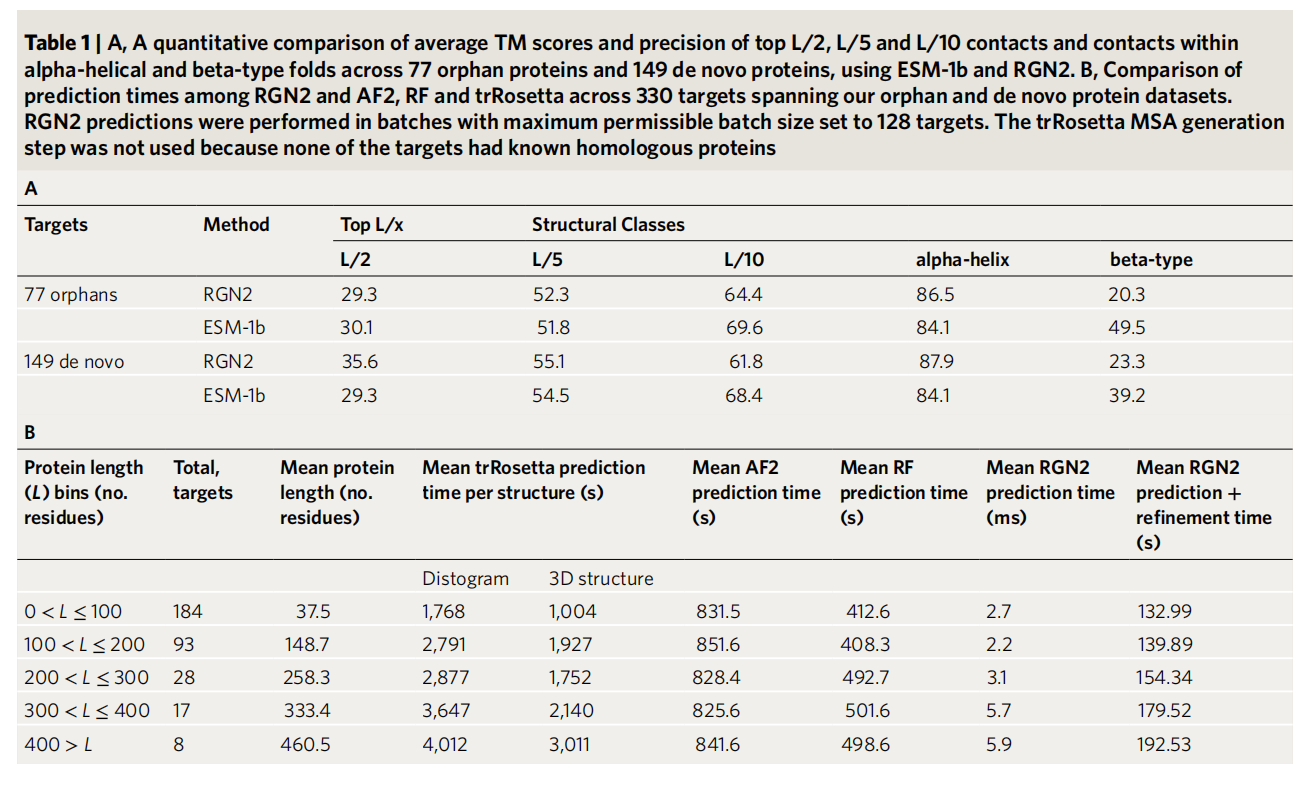
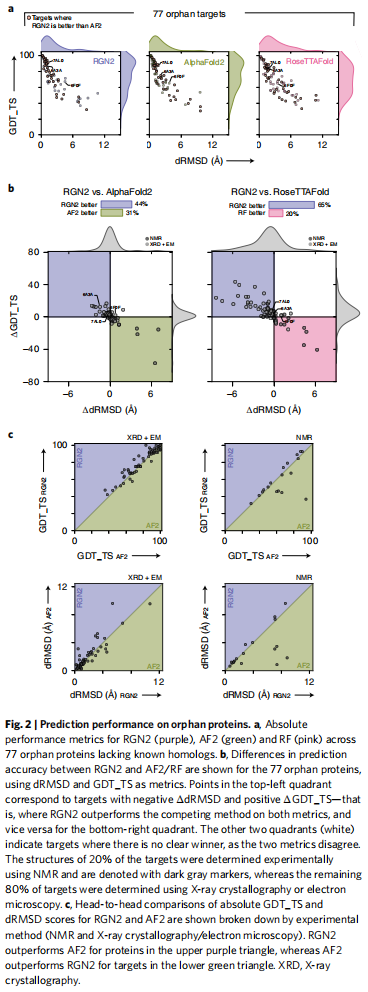
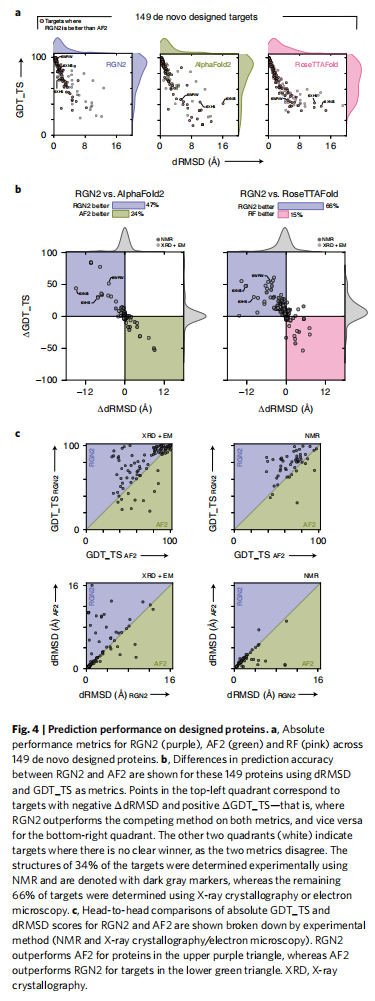
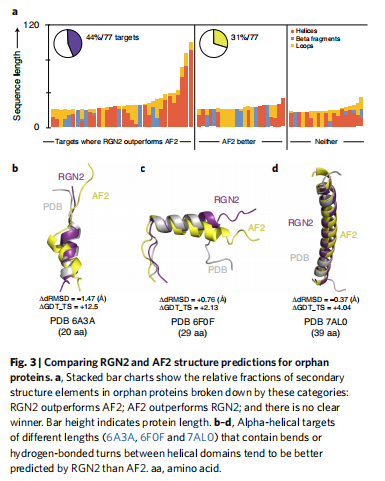
AlphaFold2和其相关的一些计算系统预测蛋白结构仍然存在一些挑战:(1)预测不能产生MSA的孤儿蛋白和快速进化的蛋白质;(2)快速探索设计的结构;(3)了解溶液中自发多肽折叠的规则。因此使用可微循环几何网络(RGN)和蛋白质语言模型(AminoBERT),平均而言,RGN2在孤儿蛋白和设计蛋白类别上的性能优于AlphaFold2和RoseTTAFold,同时使计算时间减少了10^6倍
Introduction
AlphaFold2(AF2)在最近CASP14预测挑战的部分,折叠蛋白质目标取得了高性能表明,当MSA可用,机器学习方法可以以足够的准确性预测蛋白质结构,能够与x射线晶体学,低温电子显微镜和核磁共振(NMR)等实际手段相比
但预测单条序列结构仍然是一项挑战:缺少序列同源信息时,在基因组蛋白序列~20%,在真核生物和病毒蛋白~11%,蛋白质设计和研究量化序列变体的功能和免疫性的影响仍然需要单序列结构预测
尽管对于允许使用msa的蛋白质,RGN2的性能不如基于msa的方法,但RGN2的速度却快了6个数量级
Result
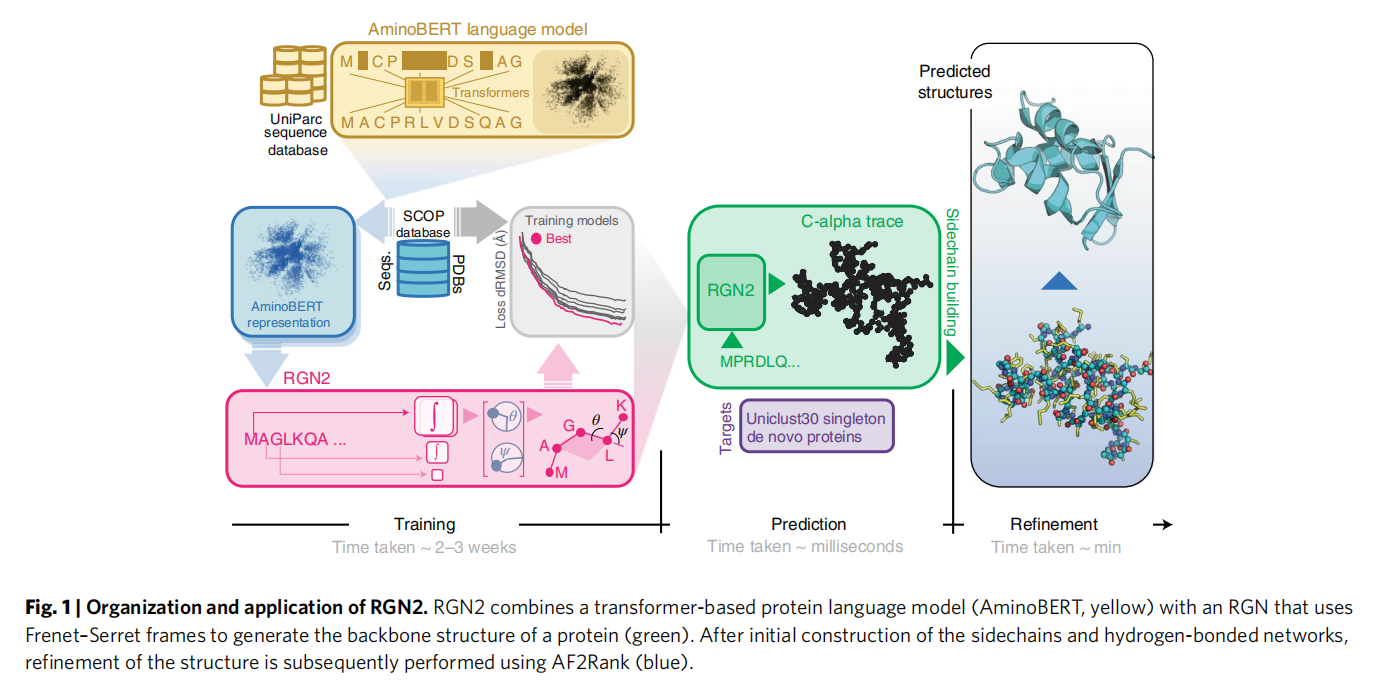
RGN2使用基于Frenet–Serret公式的方法,这种研究蛋白质几何结构的方法本质上是翻译和旋转不变的,这是溶液中多肽的一个关键特性,使用基于AF2rank的协议28改进了RGN2预测的结构,该协议推断了主链和侧链原子
-
AminoBERT:12层transformer,用从Uniparc sequence database获取的250M蛋白质序列
-
在每个序列中,同时有2-8个连续的残基被mask
-
chunk permutaion:交换连续的蛋白质片段,鼓励transformer从序列中发现整体信息
-
AminoBERT是和RGN2分别训练的,不需要微调
-
-
RNG2:
- local residue geometry是由前一帧与当前帧相关联的单一旋转矩阵描述的,与之前在RGN1中使用的扭转角相比,这种旋转和平移不变参数化的扭转角有两个优势
- 它确保了指定一个单一的生物物理参数,即~3.8A的顺序Cα−Cα距离(对应于一个反式构象),只导致物理上可实现的局部几何。这克服了RGN1的一个限制,即对一些扭转角产生了化学上不现实的值
- 通过降低了链扩展计算的计算成本的10倍,这通常主导着RGN的训练和推理时间
- 训练采用ProteinNet12数据集和一个从ASTRAL SCOPe数据集衍生出的单蛋白质结构域
- local residue geometry是由前一帧与当前帧相关联的单一旋转矩阵描述的,与之前在RGN1中使用的扭转角相比,这种旋转和平移不变参数化的扭转角有两个优势