论文地址:Learning functional properties of proteins with language models
Survey:蛋白质模型综述
Abstract
本文对蛋白质表征学习进行了详细的研究,首先对每种方法进行了分类/解释,然后对它们在预测方面的性能进行了基准测试: (1)蛋白质之间的语义相似性,(2)基于本体的蛋白质功能,(3)药物靶蛋白家族和(4)突变后蛋白质-蛋白质结合亲和力的变化。评估并讨论了每种方法的优缺点
Introduction
截至2021年5月,UniProt蛋白序列和注释知识库中约有2.15亿蛋白质条目;然而,其中只有56万(~0.26%)被专家管理员人工审查和注释,表明目前的测序(数据生产)和注释(标签)能力之间存在很大的差距,主要是由于湿实验获得结果的开销和时间很大
蛋白质功能预测(PFP)可以定义为自动或半自动地将功能定义分配给蛋白质,生物分子功能的主要术语被编纂在基因本体论(GO)系统中,PFP最全面的benchmarking project是功能注释的关键评估(CAFA),其中,参与者预测基于GO的一组目标蛋白的功能关联,其功能随后通过人工管理来识别,用于评估参与预测者的表现
在复杂计算问题中,蛋白质表征是高维的,非线性的,这就很适合深度学习技术
蛋白质表征方法可以分为两种:
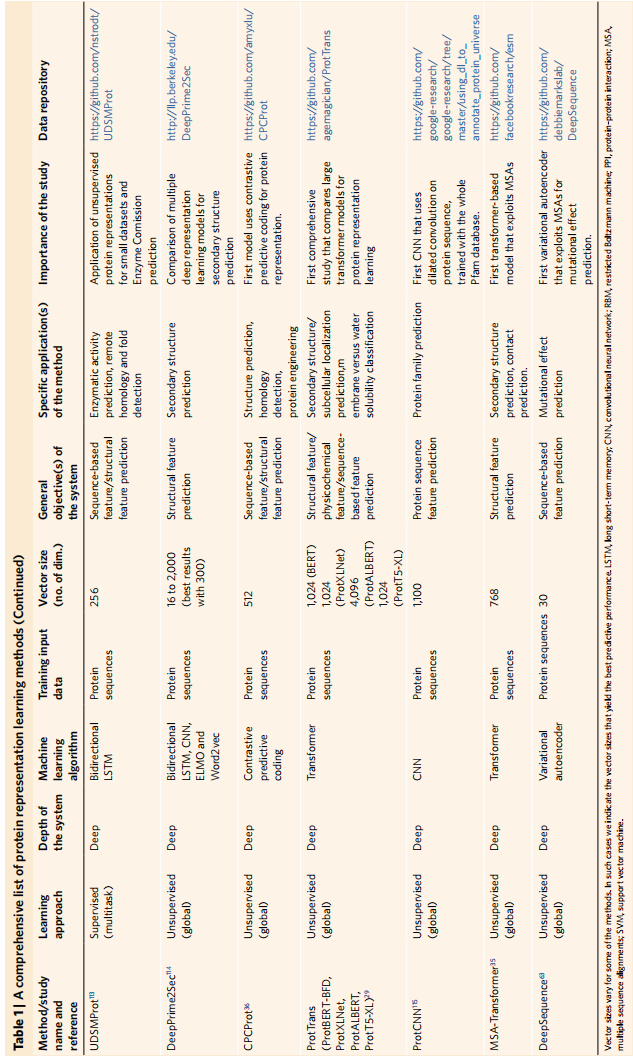
- 经典表征(模型驱动的方法):使用预定义的属性规则生成的,如基因/蛋白质之间的进化关系或氨基酸的物理化学性质,补充表1
- 数据驱动的方法:使用统计和机器学习方法通过预训练任务来训练,然后模型的输出可以用于其他蛋白质任务比如功能预测。从这个意义上说,表示学习模型利用了知识从一个任务到另一个任务的转移
目前蛋白质模型受NLP影响很深,所以也叫蛋白质语言模型
在本研究中,对自2015年以来提出的现有蛋白质表示学习方法进行了全面的调查,并通过详细的基准分析来测量这些方法在捕获蛋白质功能特性方面的潜力,包含了经典方法和人工智能方法
为了评估每个表示模型捕获功能信息的不同方面的程度,我们构建并应用了基于以下条件的基准;(1)蛋白质之间的语义相似性推理,(2)基于本体的PFP,(3)药物靶蛋白家族分类,(4)蛋白质-蛋白结合亲和度估计
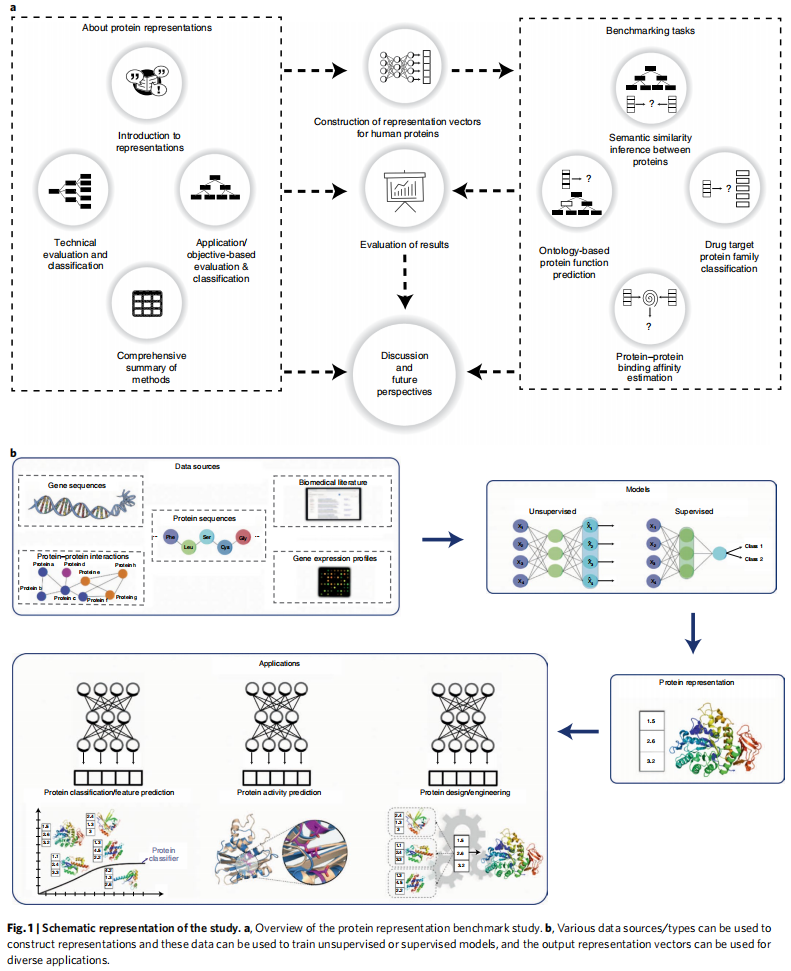
整个研究总结如图1a
作者提供了一个Benchmark软件(PROBE)
Results
蛋白质表征的构建和应用在fig1b
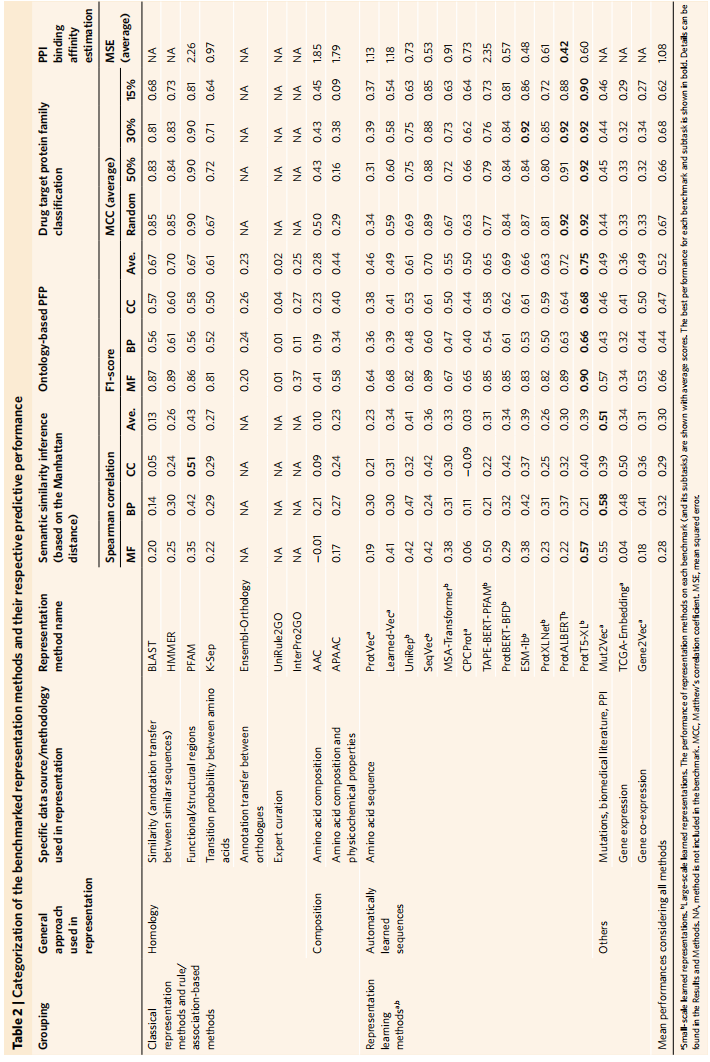
所有方法在四个基准测试上的平均性能在表2中
根据预测属性的分辨率,蛋白质表示学习方法可分为蛋白质级或残基级特征的两类之一,主要关注前者
Semantic similarity inference
该分析旨在测量多少信息表示模型捕获了多少关于生物分子功能相似性
首先使用余弦、曼哈顿和欧氏距离/相似性计算了数据集中蛋白质表示向量之间的成对定量相似性,然后与这些蛋白质的ground truth相似度进行比较,就是相同蛋白质对的表示向量相似值和实际基于GO的语义相似值之比
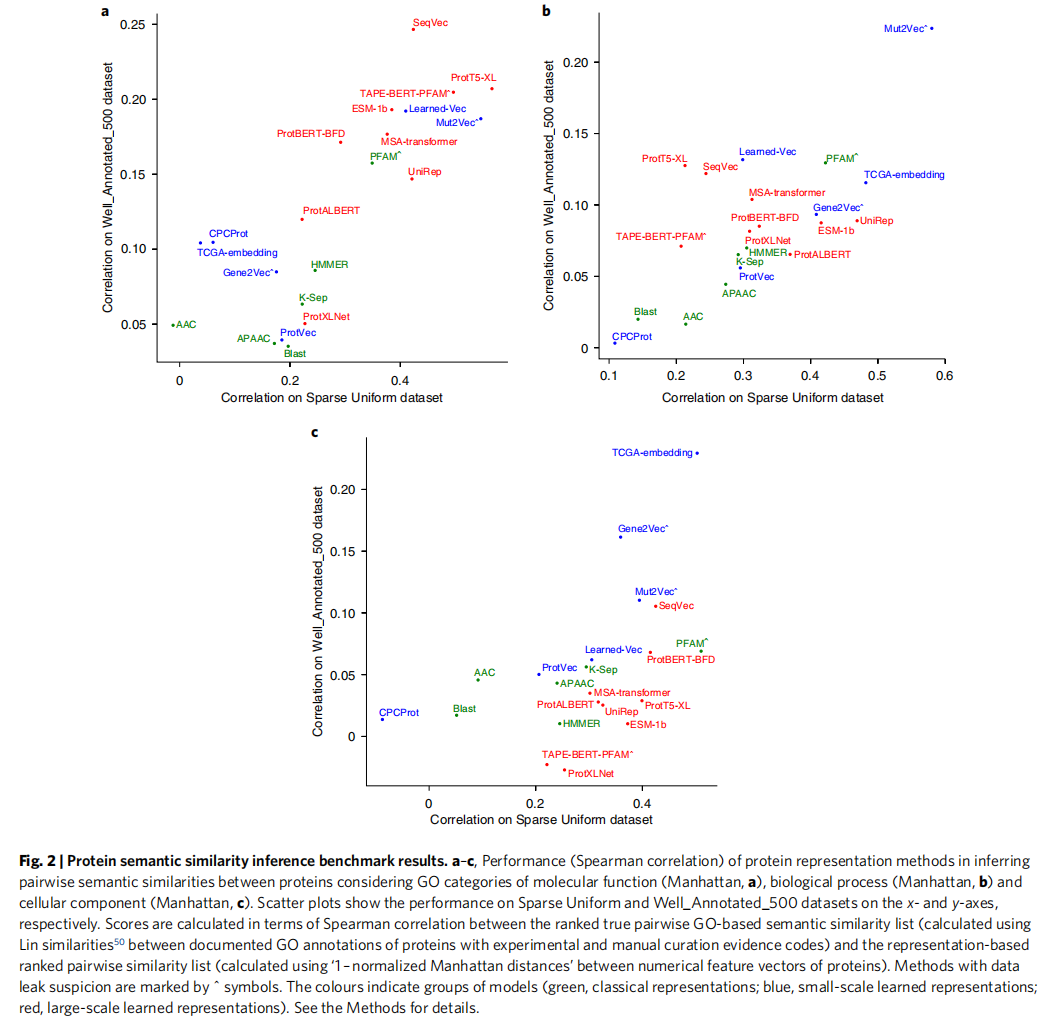
从图2a可以看出ProT5-XL在GO molecular function(MF) 是最成功的表征;Mut2Vec在GO biologica process(BP) 最好;TCGA_EMBEDDING和PFAM在 GO cellular component (CC)最好
构建我们的基准时,我们首先使用整个参考人类蛋白质组作为我们的测试数据集;然而,~20.000蛋白质之间的所有成对组合被证明是一个稀疏比较空间,使得测试方法之间的差异在统计学上不显著。除了数据之外,还有一个很重要的超参是指标,作者试了余弦,曼哈顿,欧几里得等,应该综合不同指标下的结果避免造成误解
Ontology-based PFP
评估基于分类的自动PFP中表示模型的成功程度
在这个基准下用了一个线性分类头(来自scikit-learn的具有随机梯度下降(SGD)优化器的线性支持向量分类)
图3中基于f1分数的热图给出了9个GO组(low、middle、high]×[shallow、normal、specific])的PFP性能结果
可以看到效果要比CAFA challenges好,因为只跑了一个测试样本而不是所有测试样本,实验设计选择是为了防止所有基准方法在低性能区域的分数积累(特别是对于BP等难以预测的本体),这将阻止性能的清晰比较。我们的目的是在一个高度控制的环境中从不同的角度相互比较这些方法,而不是找到PFP的最佳总体方法,这是CAFA challenges的目标
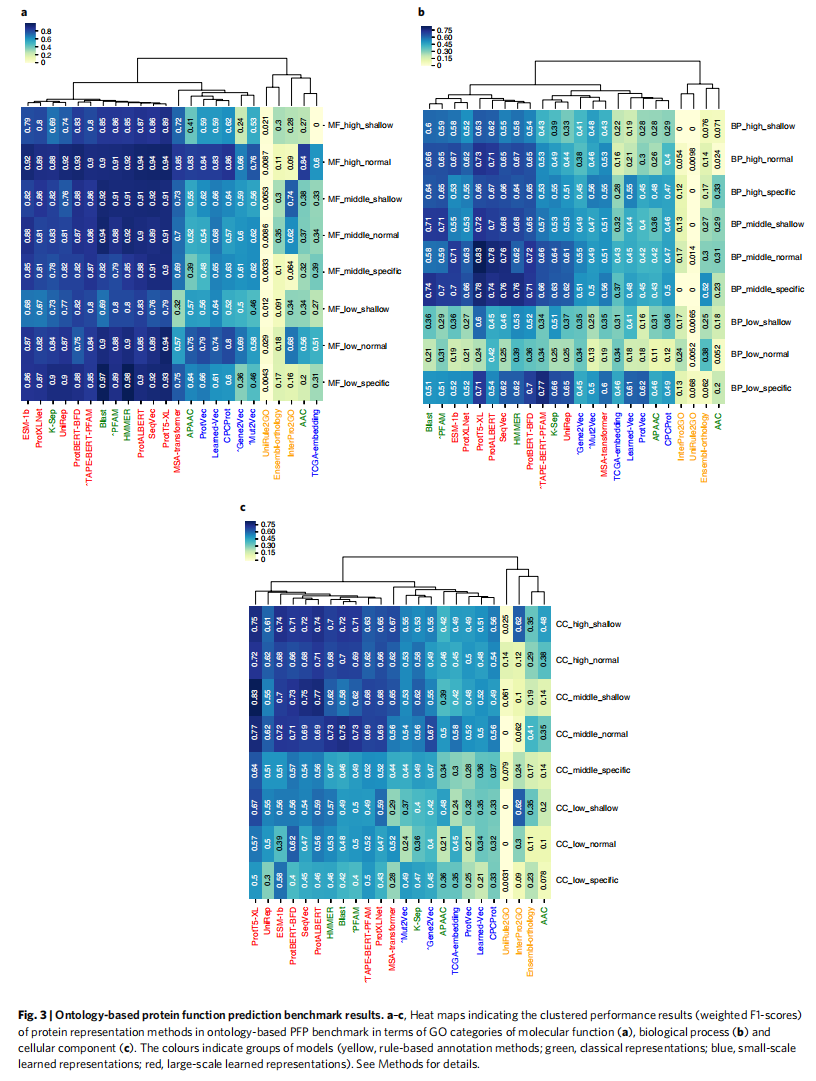
从图3看出最好的方法都差不多,PortT5-XL在MF,BP和CC都是最好
CC和BP预测任务总体表现低于MF预测任务,这是合理的,因为大多数基于学习的方法使用蛋白质序列数据作为输入,而且该序列不是定位的直接指示物(因为被切割的信号肽缺失),也不是蛋白质在大规模过程中的生物学作用
CC项预测的成功率随着注释蛋白数量的减少而降低,BP和MF也能看到相似的现象。但是增加或减少术语特性(即浅/通用术语与特定/信息术语)没有观察到类似的现象,可能指出,对于特定/信息丰富的GO术语的预测仍然存在一个问题,因为其中许多术语的注释蛋白数量较少
Drug target protein family classification
在药物发现的框架下测量了蛋白质表征的性能,通过预测药物靶蛋白的主要家族(即酶、膜受体、转录因子、离子通道等)
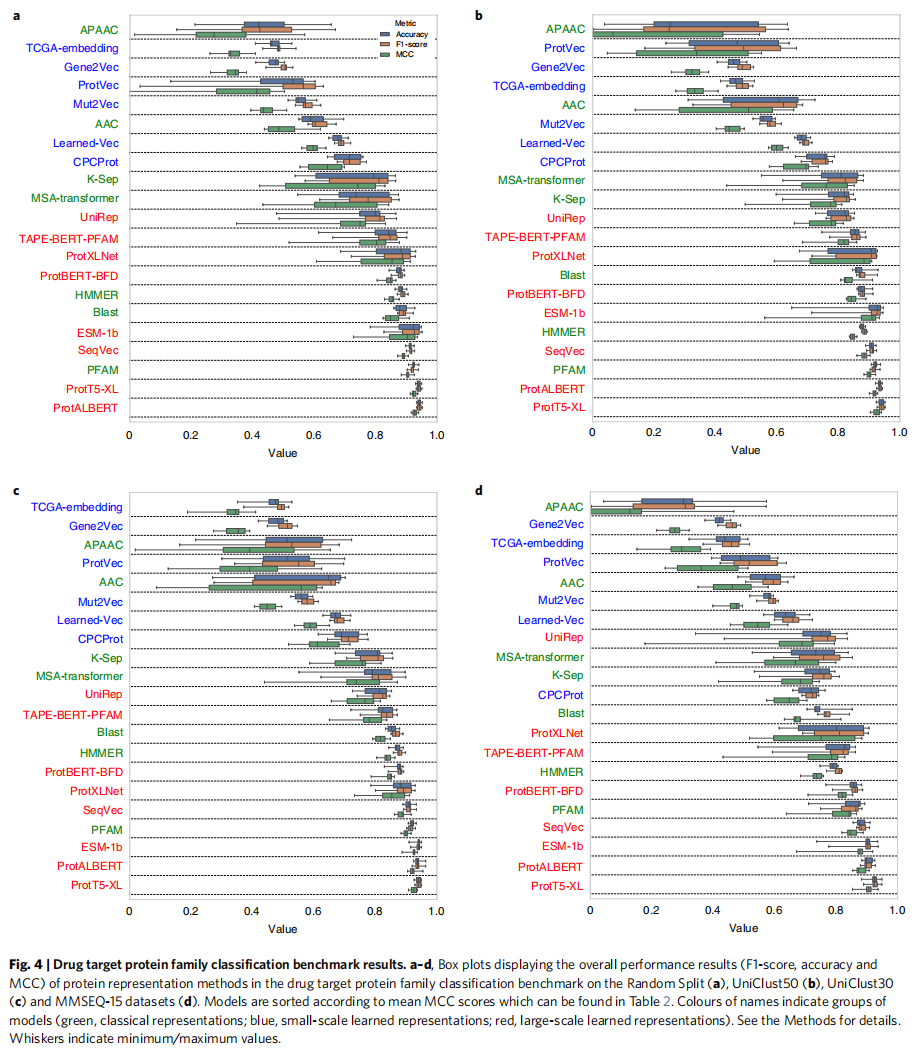
制备四种不同版本的蛋白质家族注释数据集,每一个都根据不同的预定序列相似性阈值(即,随机分割数据集,以及分别使用Uniclust50、Uniclust30和MMSEQ-15聚类的50%、30%和15%相似性阈值数据集)进行过滤,以用于十折交叉验证分析中的训练/验证数据集分割
根据相似性设阈值可以检查模型是简单的学序列的相似性还是学习的是预测任务背后复杂的隐式模式
可以看出PortT5-XL还是最好的,同时地相似度的情况下性能都在普遍下降,但经典方法比表征学习方法下降的更明显。当相似性阈值降低到15%,甚至低于所谓的蛋白质之间传递结构和功能注释的黄昏区(即~25%的序列相似性)时,基于顶级表示学习的方法仍然表现得很好,这表明表示学习方法可能有能力捕获简单序列相似性之外的模式
Protein–protein binding affinity estimation
评估了表征方法在预测实验确定的蛋白质-蛋白质结合亲和力方面的性能
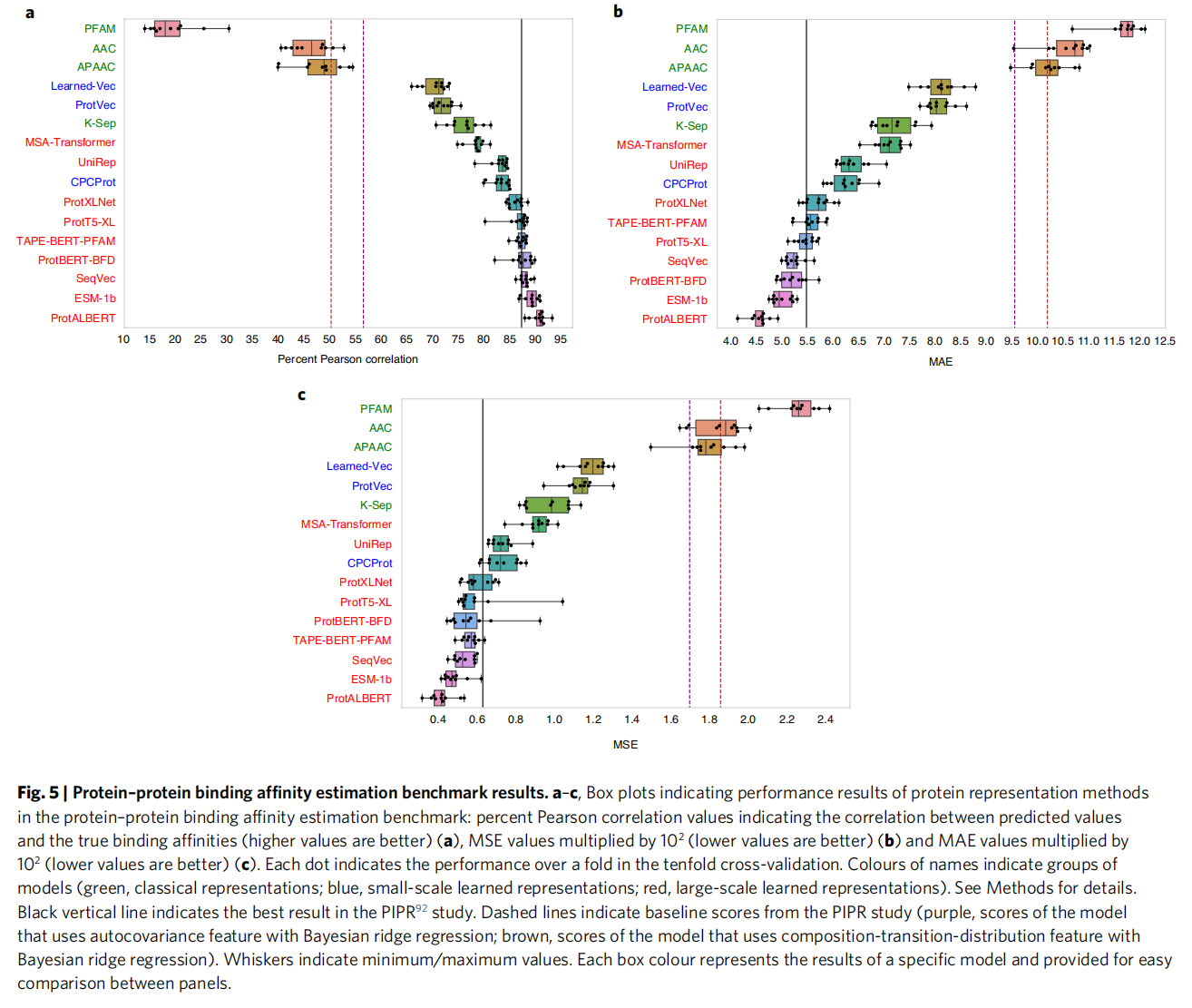
PortALBERT是最好的
PIPR在有监督的框架中学习输入序列(以端到端的方式),旨在最大化结合亲和力预测性能。相反,我们的基准中的蛋白质表示是通过与结合亲和力预测完全无关的任务(例如,预测序列中的下一个氨基酸)学习的(在预训练期间),然后通过简单回归以监督的方式对结合亲和力值进行训练
这可以通过注意力机制来解释,PortTrans显示注意力头可以获取氨基酸之间的互相作用,这就能解释为什么ProtALBERT效果最好,这个模型比其他transformer方法参数少,但是注意力头最多
Discussion
蛋白质表征方法是表1,在四个预测基准上的整体表现是表2
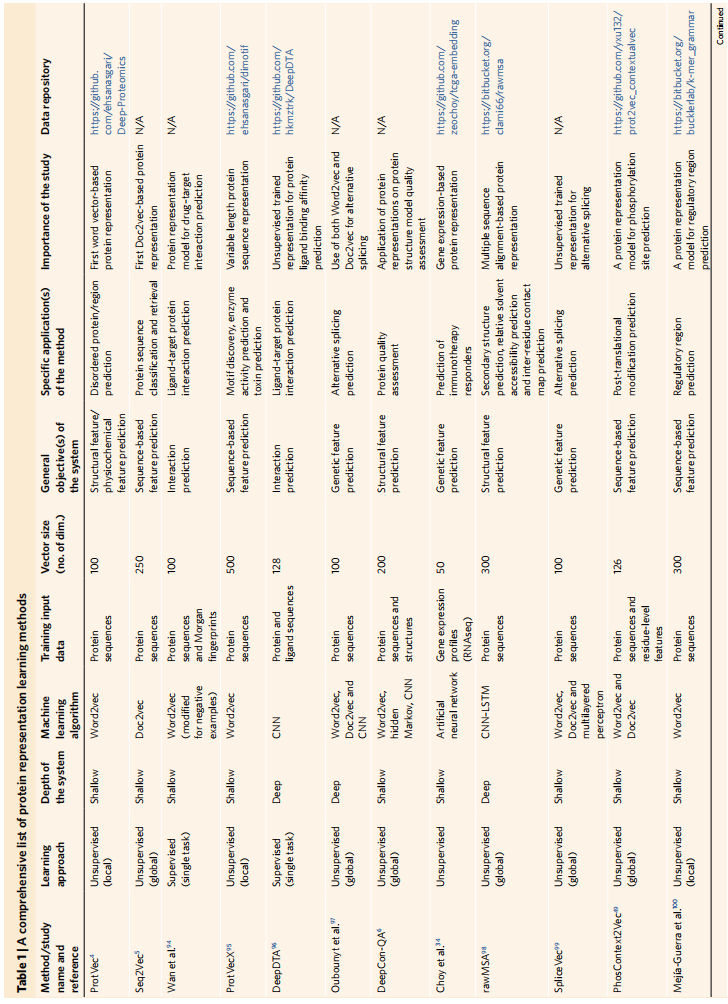
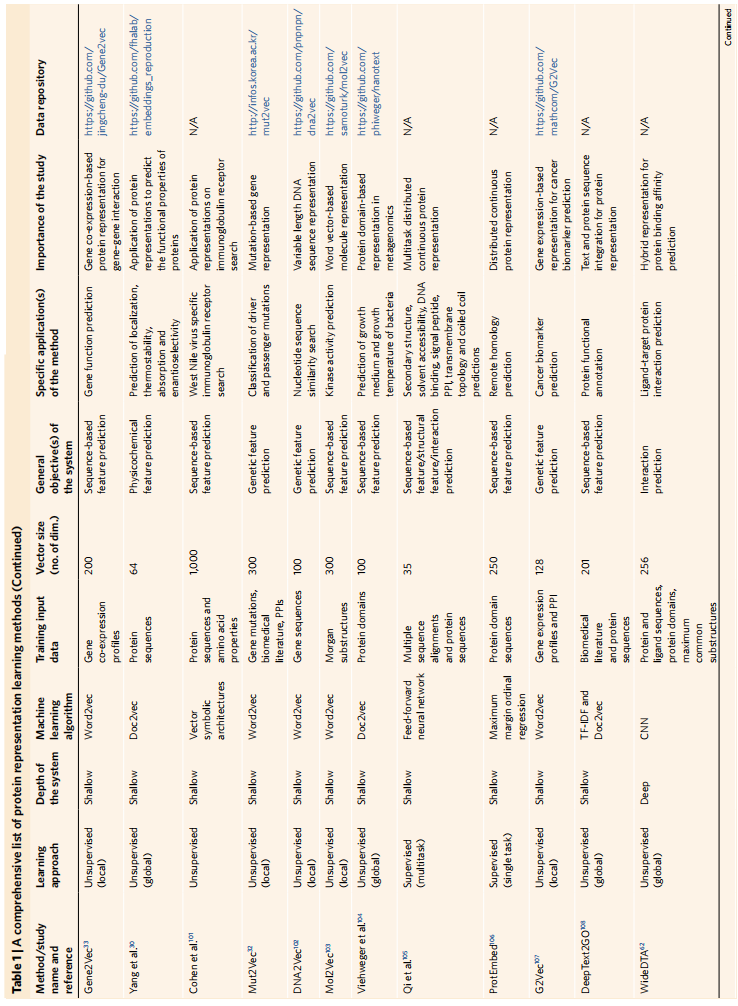
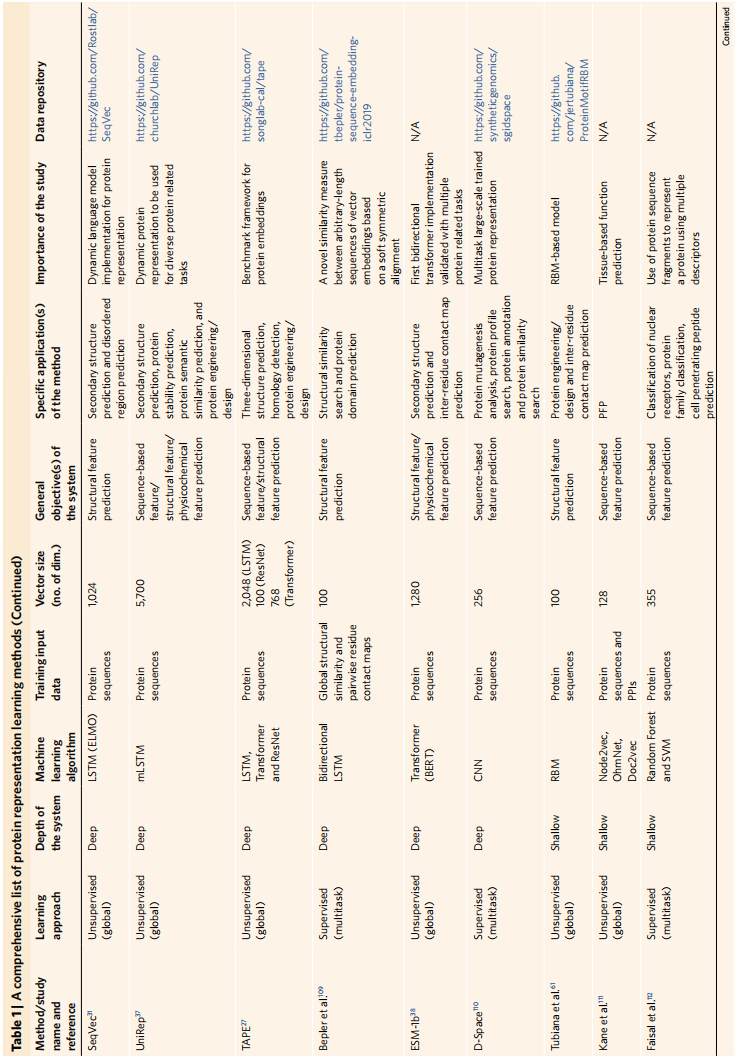
结论:
- 基于表示学习的方法在蛋白质的功能分析中通常比经典的方法表现得更好
- 模型设计和训练数据类型/源是表示学习的关键因素
- 比如表2里面的两种BERT,TAPE-BERT-PFAM用3200万个蛋白质结构域序列进行训练。ProtBERT-BFD用21亿个宏基因组序列片段进行训练;然而,这两者之间的性能差异不显著
- 同时更复杂的模型比如PortT5-XL用同一个21亿数据有更好的表现,所以模型架构设计是最重要的
- 训练多种数据类型表现有提升
- 在蛋白质表示学习方法的构建和评估时,应考虑潜在的数据泄漏
- 蛋白质表征学习的现状和挑战
- 虽然起源于NLP,但蛋白质和语言还是有结构差异的
- 蛋白质词表小,蛋白质表征中较低数量的构建块(即20个氨基酸)可能是较小模型在蛋白质表征学习领域与较大模型竞争的优势,所以鼓励蛋白质特有的学习模型
- 模型可解释性对理解模型表现很关键,但本文提到的大多数模型都是不可解释的。数据科学领域通常会想把真实的属性和输出向量的每个位置联系起来
- 蛋白质表征模型只用了一个类型的数据(序列),但实际上蛋白质有很多不同的生物信息比如PPIs、翻译后修饰、基因/蛋白(co)表达等
- 像Mut2Vec用了PPIs、突变和生物医学文本获得了更好的准确度;
- MSA-Transformer和无向图模型(比如DeepSequence)利用了同源信息,虽然DeepSequence使用MSA的后验分布来计算潜在因素,但MSA Transformer使用基于行和列的注意力来组合MSA和蛋白质语言模型。MSA Transformer能够捕获进化关系
- 提出可能有两种方法能够获得更完整的表征:
- 把不同类型的生物数据独立编码后concat起来可能可以得到更完整的表征,然后训练融合这些向量
- 使用异构图来学习
- 蛋白质表示学习方法可用于设计新的蛋白质
- 探索的序列空间大:人类的平均蛋白质有350个氨基酸,就会有20^350个组合,即使大多是无意义的
- 生成模型可以从真实样本中学习概率分布并合成样本
Methods
根据应用领域形成了五个主要类别:(1)蛋白质相互作用预测(对于理解分子机制和途径至关重要),(2)物理化学特征预测(对于蛋白质工程和药物发现相关任务很重要),(3)遗传特征预测,(4)PFP和(5)结构特征预测
Semantic similarity inference benchmark
下载了UniProtKB/Swiss-Prot数据库中的所有人类蛋白质条目以及UniProt GOA数据库中的GO术语注释(2019_11版本)。标有IEA证据代码的电子推断注释被排除在数据集中,只剩下人类专家审查的注释。随后根据真实路径规则,通过将注释传播到GO图中断言项的父项来丰富数据集
最终的完整注释数据集包含14625个不同的GO术语(其中3374个属于MF,9820个属于BP,1431个属于CC)和326009个注释(其中75884个属于MF,154532个属于BP和95593个属于CC)
用GoSemSim包里的lin similarity计算相似度
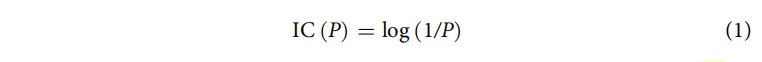
LCS是两个GO项在GO有向非循环图中的根时的第一个共同祖先
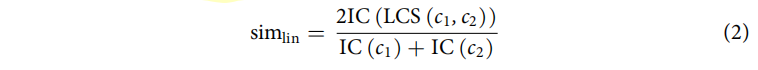
3077个蛋白质用于计算基于MF的成对语义相似性,6154个蛋白质用于基于BP的相似性,4531个蛋白质用于CC的相似性,但里面有很多是低注释质量的蛋白质,所以做了子数据集 (Well_Annotated_500, Well_Annotated_200 and Sparse Uniform)
- 第一个子集,仅包含按GO注释数量排序的前500个蛋白质(在相关图中标记为注释良好的500个)
- 第二个子集仅由前200个这样的蛋白质组成(在相关图中标记为Well_Annotated_200)
- 在上述三个数据集中,相似性分布不均匀,产生了非常密集的相似性得分区域(补充图2),由于具有近相似性的对之间的秩变化,显著降低了相关值。这导致低相关值周围的累积,降低了测量的辨别能力。为了防止这种情况,我们从注释良好的500集合中的成对相似性排序列表中抽取每千个蛋白质对,以生成均匀分布的数据集。该最终数据集包含40种不同蛋白质之间的247个相似性得分(在相关图中标记为稀疏均匀)。因此,在我们的三个数据集中,稀疏一致性是最容易预测的,而Well_Annotated_500是最具挑战性的
这样就有两组成对相似度数组:第一个是通过考虑我们数据集中蛋白质之间GO衍生的语义相似性(即ground truth语义相似性)来计算的,第二个是由直接从表示向量计算的成对相似性组成的
Ontology-based PFP benchmark
对于每个GO类别(即MF、BP、CC)的数据处理:
- 从UniProtKB/Swiss-Prot和UniProtGOA数据库中获得了人类蛋白质及其GO术语注释(两者均发布2019_10)
- 从GO术语注释列表中排除了所有电子注释(证据代码:IEA),目的是提高注释的可靠性并防止预测过程中的错误传播
- 对于每一个GO术语创建了一个单独的列表,用于通过交叉验证进行模型训练和测试。使用UniRef cluster筛选每个蛋白质列表,UniRef提供了基于序列相似性形成的蛋白质簇。使用UniRef50聚类,每个列表中没有序列相似性超过50%的蛋白质序列
- GO术语根据注释蛋白的数量分为low、middle或high,2-30是low,100-500是middle,1000以上high
- GO术语专有性分为shallow、normal和specific,各分支最大深度的前三分之一内的术语被认为是shallow,第二个三分之一是normal,最深的三分之一是specific
- 上面的两种三个分组组合成9个,加上三种GO类别就是27组,但有两类是没有的(MF-high-specifc and CC-high-specifc),所以只分析25组
Drug target protein family classification benchmark
使用了ChEMBL数据库(v.25)54,该数据库包含药物/化合物-靶蛋白相互作用数据(即生物活性)的精选集,用于药物发现和开发的实验和计算研究,使用了四个广泛的靶蛋白家族,并将其余靶蛋白分为第五类(即酶、膜受体、转录因子、离子通道和其他)
将数据分成不同相似程度的训练和测试数据集,构建了四个数据集。使用基于蛋白质序列相似性的聚类方案UniClust,预先计算的不同粒化水平(如50%、30%)的序列聚类,还为人类蛋白质创建了另一个粒化水平为15%的簇
Protein–protein binding affinity estimation benchmark
使用的是突变蛋白相互作用动力学和能量学结构数据库(SKEMPI)数据集
在基准测试阶段,我们使用SKEMPI中的2950个数据点作为训练/测试数据集,测量了蛋白质表示方法在直接预测结合亲和力值(包括属于野生型和相互独立的突变蛋白的测量)方面的性能。

