论文地址:ProtTrans:Towards Cracking the Language of Life’s Code Through Self-Supervised Learning
ProtTrans:基于transformer的不同蛋白质模型
Abstract
根据UniRef和BFD的数据训练了两个自回归模型(Transformer XL、XLNet)和四个自编码器模型(BERT、Albert、Electra、T5),这些模型包含多达3930亿个氨基酸。LM在Summit超级计算机上使用5616个GPU和最高1024个核心的TPU Pod进行训练。在后续一些任务上用embedding作为唯一输入验证了有效性,第一个是蛋白质二级结构的每残基预测(三态精度Q3=81%-87%);第二个是蛋白质亚细胞定位的每蛋白质预测(十态准确度:Q10=81%)和膜与水溶性(二态准确度Q2=91%)。对于残基预测而言,使用embdding(ProtT5)首次超过了SOTA而没有用进化信息避开了昂贵的数据库搜索
Introduction
计算生物学里最好的预测方法是结合了机器学习(ML)和进化信息(EI)
- 首先,搜索总结为多序列比对(MSA)的相关蛋白家族,并提取该比对中包含的进化信息
- 第二,通过监督学习内隐结构或功能约束将EI输入ML
但使用EI也有缺点:
- 当预测整个蛋白质组的时候,编译所有的蛋白质EI计算昂贵
- EI不是所有蛋白质都有
- 当EI多样化很好时效果提升最好
- EI的预测是依靠家族,可能无法区别同一家族中的不同蛋白质
- AlphaFold2,基于EI和ML的高级组合;尽管该方法以前所未有的精度预测蛋白质3D结构,但AlphaFold2模型的计算成本比EI的创建高出许多数量级
首先,探索了在蛋白质上训练的大规模语言模型以及用于训练的蛋白质序列数据库的局限性
其次,比较了自回归和自动编码预训练对后续监督训练成功的影响,并使用进化信息(EI)
Methods
Data for protein Language Models (LMs)
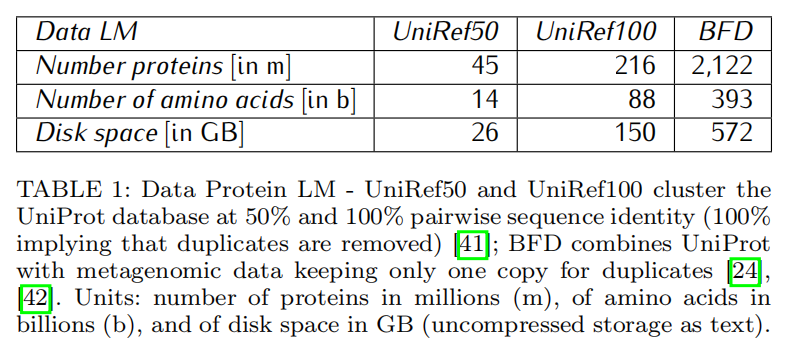
BFD数据集最大,但UniRef100比BFD长1.6倍
Embeddings for supervised training
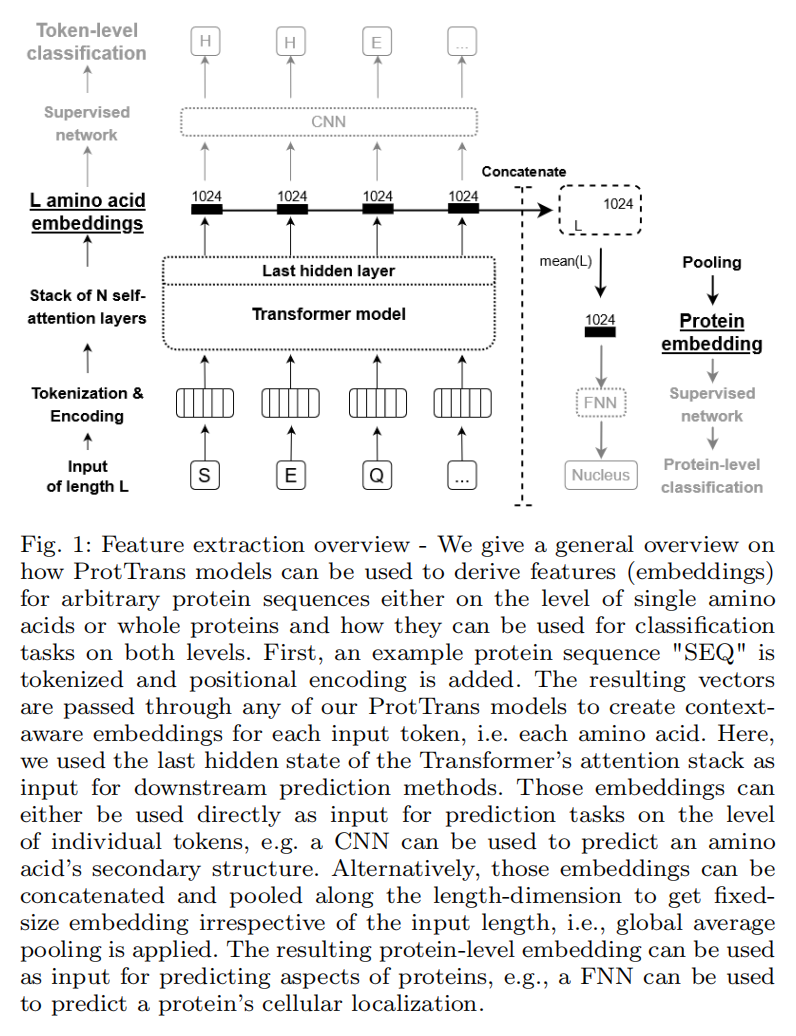
用embedding来做有监督的迁移学习
Per-residue prediction/single tokens
包括其他公共测试数据集,即CB513、TS115和CASP12。这些都有局限(CASP12:太小,CB513和TS115冗余且过时)。因此,我们添加了一个新的测试集,仅使用NetSurfP-2.0发布后(2019年1月1日之后)发布的蛋白质。包括PDB中的蛋白质≤ 2.5Å和≥ 20个残基。具有最高灵敏度(-7.5)的MMSeqs2去除了对训练集或自身具有>20%PIDE的蛋白质。在顶部,PISCES去除了其程序认为具有>20%PIDE的任何蛋白质。这些过滤器将新蛋白质(链)的数量从18k减少到364(称为集合NEW364)
Per-protein prediction/embedding pooling
对蛋白质特性的预测类似于NLP里面句子分类,用的DeepLoc数据集
Data for unsupervised evaluation of embeddings
通过使用t-SNE将高维表示投影到二维(2D),评估了从蛋白质模型中提取的嵌入所捕获的信息
Step 1: Protein LMs extract embeddings
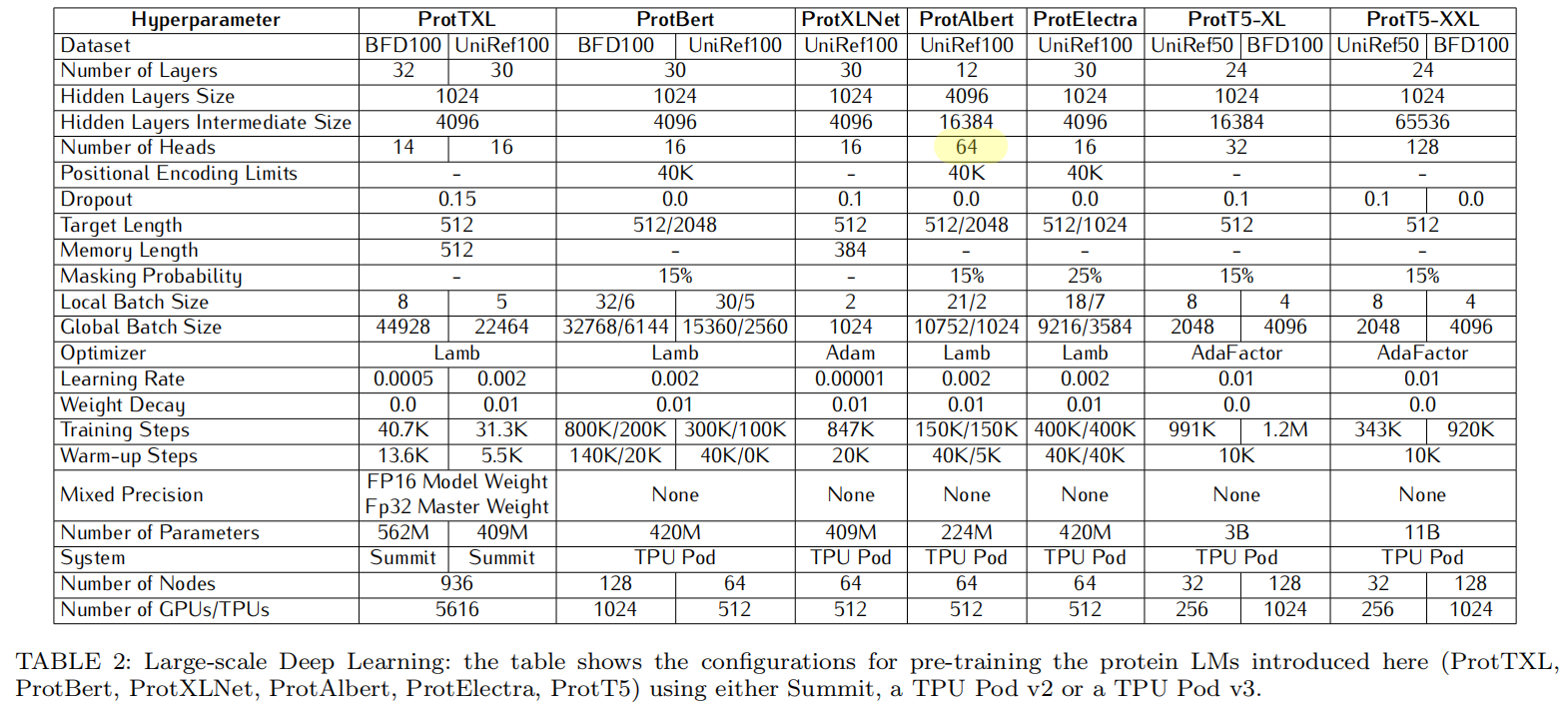
ProtAlbert用了64个注意力头
Step 2: Transfer learning of supervised models
per-residue prediction
把embedding丢入两层CNN,第一层用大小为7的窗口把embedding压缩到32位,压缩后的表征输入两个CNN(窗口为7),一个学预测三态的二级结构,一个学八态,多任务学习
per-protein prediction
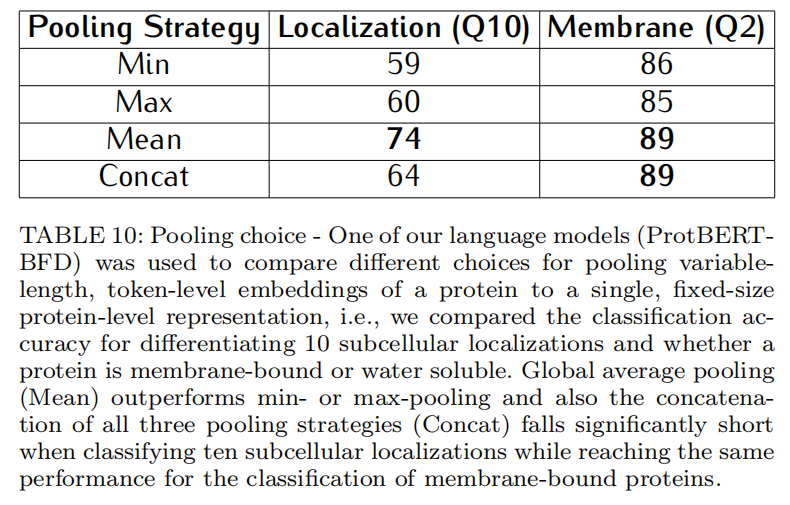
把embedding先做池化,不同池化策略表10,选择对所有实验用mean-pooing,输入一个32个神经单元的层,同时预测亚细胞定位和膜结合蛋白和水溶性蛋白之间的分化(多任务学习)
Software
NVIDIA APEX用于混精度,支持Pytorch,NV-LINK对ProtTXL减少了60%时间,对ProtBert减少了72%时间
Resluts
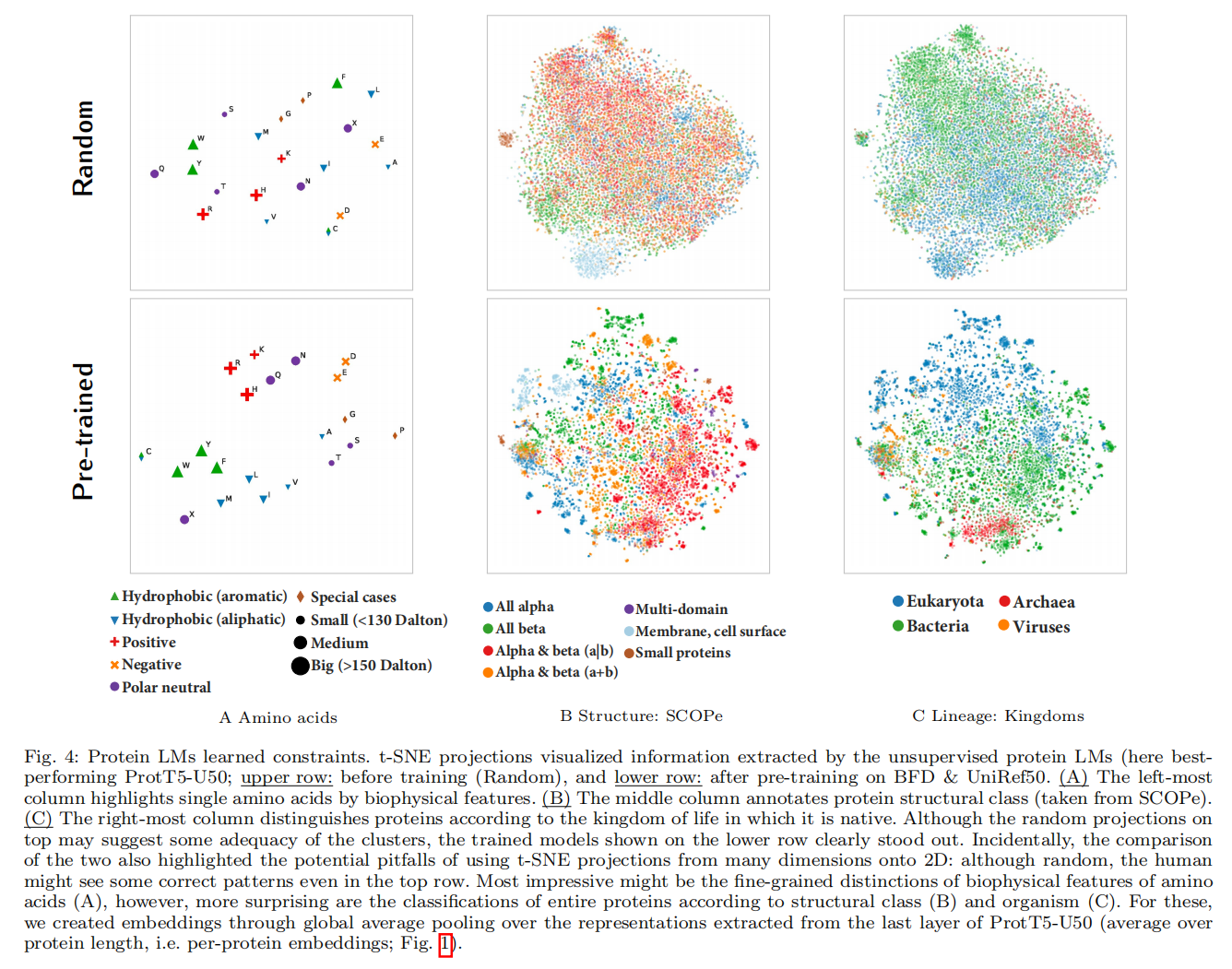
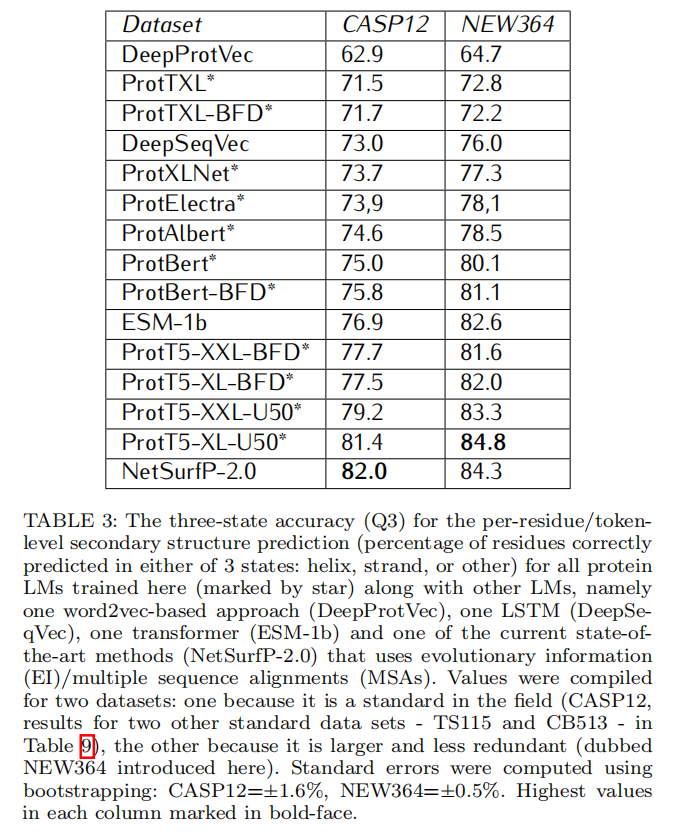
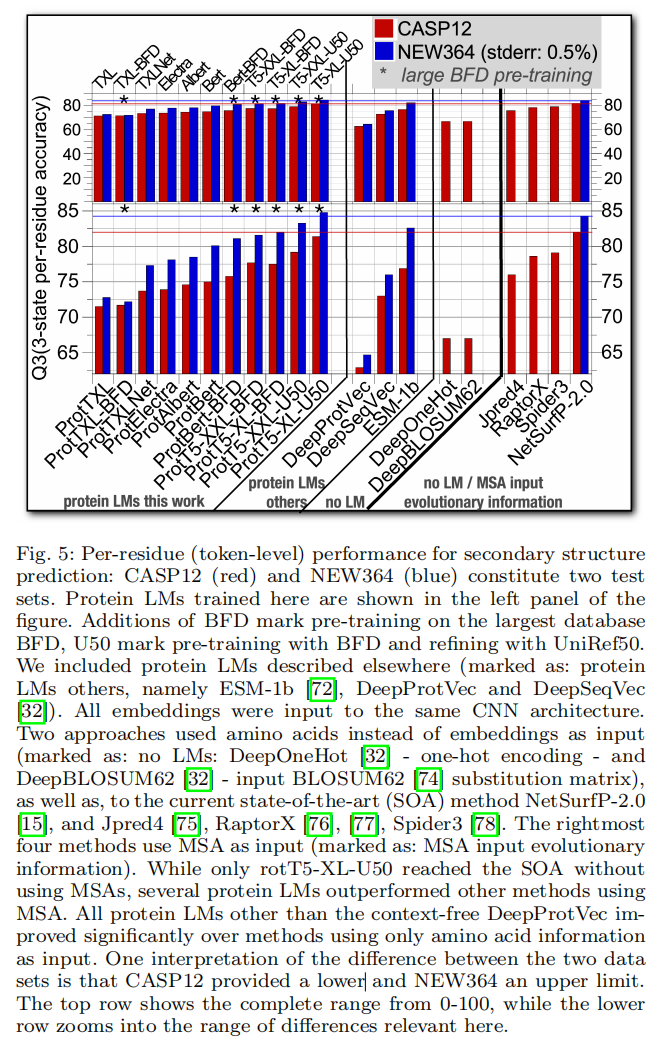
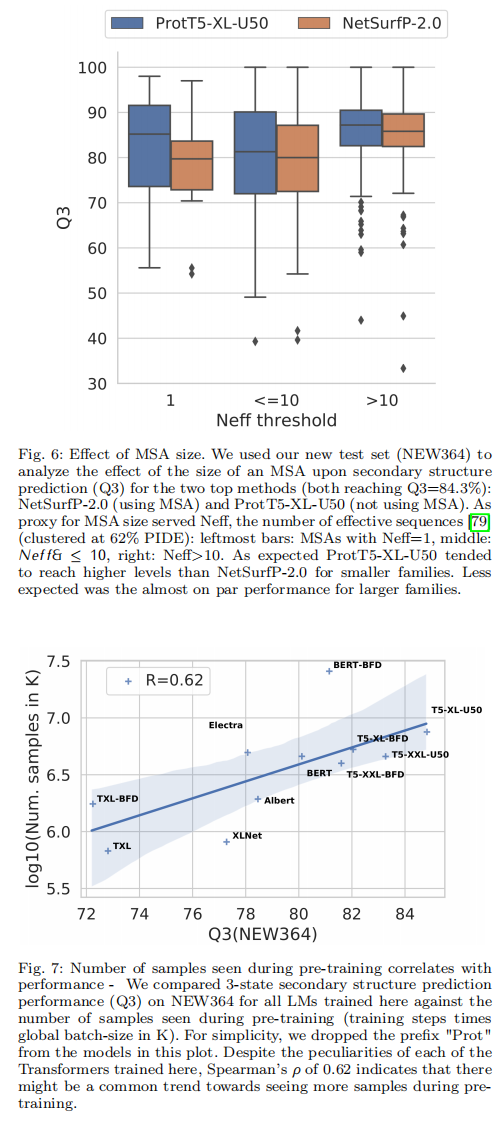
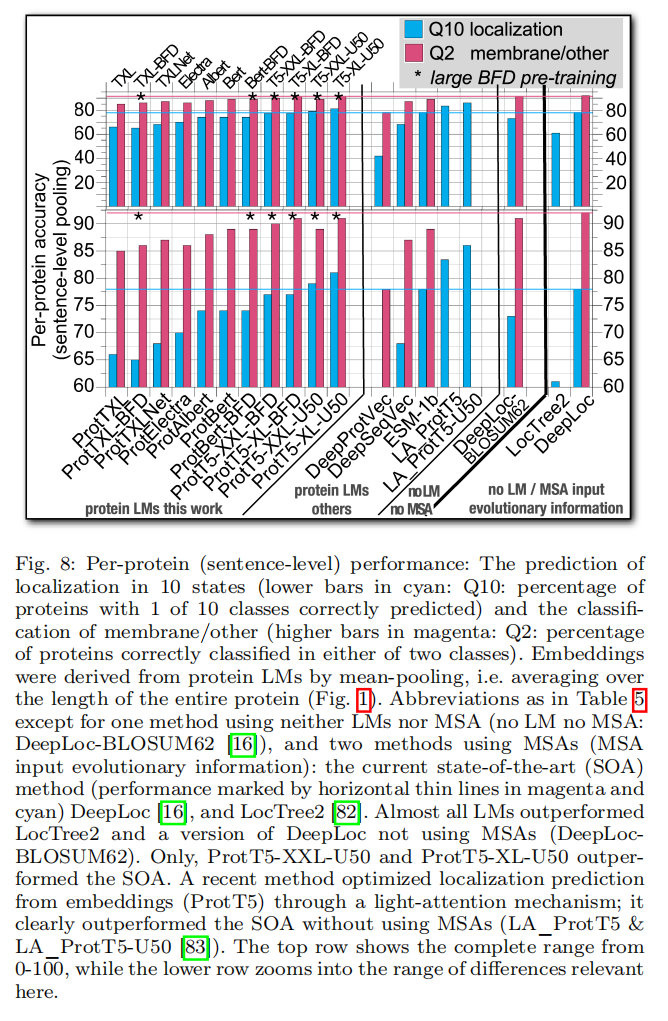
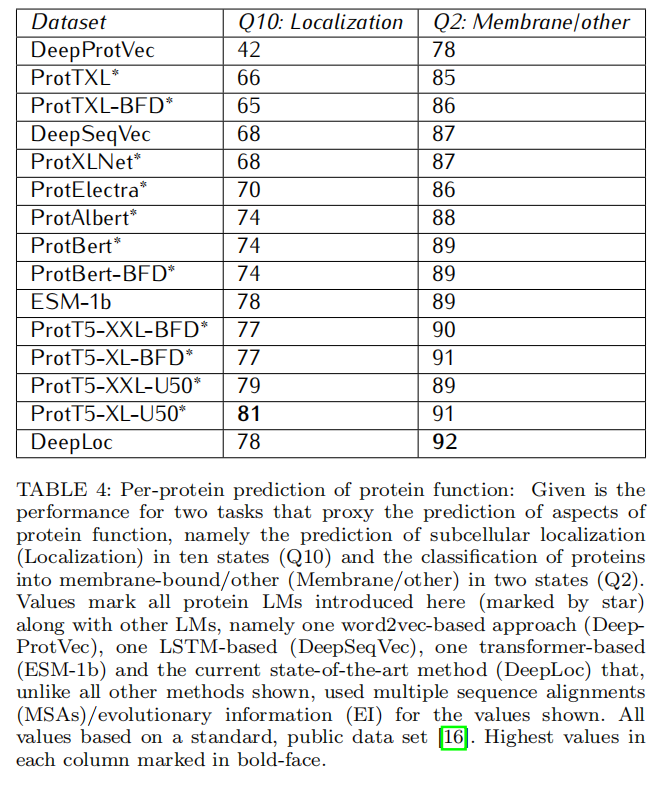
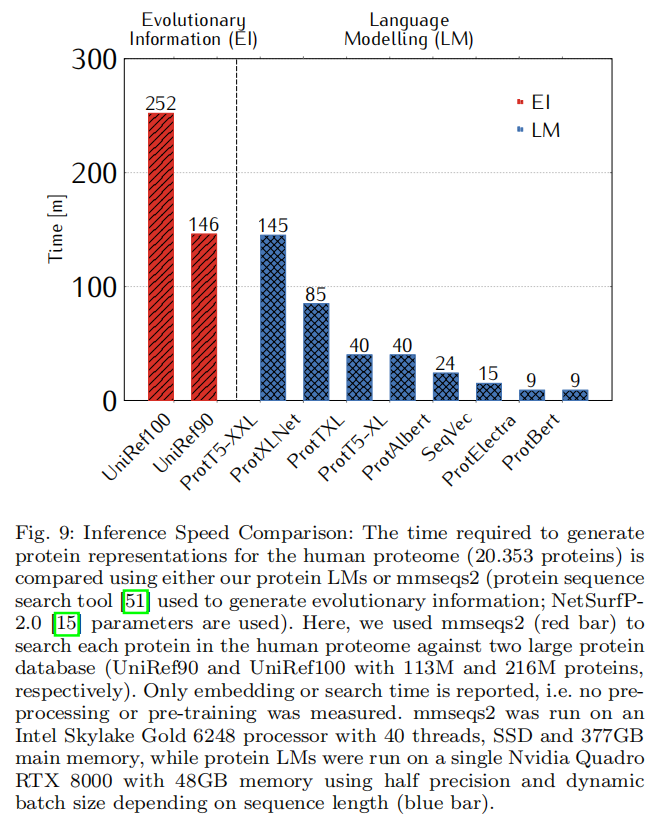
Discussion
Have protein LMs reached a ceiling?
(1) 噪声较小且冗余度较低的语料库(例如UniRef50)比噪声较大但冗余度较高的语料库(如BFD)有所改善
(2) 从我们有限资源的角度来看,使用资源进行足够长的训练是最重要的,因为在预训练期间看到的样本数量与下游任务的预测性能相关。最终,这似乎源于足够的模型大小和样本吞吐量之间的权衡
(3) 双向模型优于单向模型。然而,鉴于在这项工作的回顾过程中蛋白质LMs的进展,我们还没有看到任何证据表明蛋白质LMs达到了极限
许多开放性问题:
(1) BERT或Albert提供的下一句话或句子顺序预测等辅助任务的添加是否适合蛋白质序列?一种建议可能是使用结构信息或进化关系
(2)transformer蛋白LM训练的效率是否可以通过稀疏transformer或使用局部敏感哈希(LSH)优化注意力来提高,如Reformer模型[99]或线性transformer的最新工作所介绍的?
(3) 哪些数据集预处理、减少和训练批采样应最佳使用,以获得更好的结果?
(4) 根据特定任务定制受监督的培训管道将提高多少?我们将二级结构或定位预测更多地作为展示蛋白质LMs成功的代理,而不是作为一个独立的终点
(5) EI和AI的结合会带来未来最好的蛋白质预测吗,还是单蛋白质预测的优势(速度、精度)会胜出?事实上,单蛋白预测还具有更精确的优势,因为它们不提供蛋白质家族的一些隐含平均值

