论文地址:Masked inverse folding with sequence transfer for protein representation learning
Model code is available at https://github.com/microsoft/protein-sequence-models. Pretrained model weights and AlphaFold predictions used in this work are available at https://doi.org/10.1234/mifst
MIF:利用预训练语言模型帮助蛋白质结构预训练
Abstract
sequence-only方法忽视了实验和预测得到的蛋白质结构信息,inverse folding方法根据蛋白质的结构重建蛋白质的氨基酸序列,但不利用没有已知结构的序列。本项工作训练一个masked inverse folding蛋白质语言模型参数化为一个结构图神经网络。并证明了使用预先训练的序列蛋白质掩蔽语言模型的输出作为逆折叠模型的输入,进一步改善了训练前的困惑
Introduction
使用inverse folding作为预训练的任务,因为直觉上认为整合结构信息应该会提高下游任务的性能。现有的inverse folding方法还是必须在序列已知或预测出有结构的情况下训练,没有用上大量的没有接的序列数据,比如UniRef50有42m序列,但PDB只有190k实验验证的结构
用GNN训练一个掩码语言模型称作MIF,使用预训练好的sequence-only蛋白质MLM作为MIF输入能够改善预训练的困惑度,这种模型称为MIF-ST(Masked Inverse Folding with Sequence Transfer)
MIF and MIF-ST
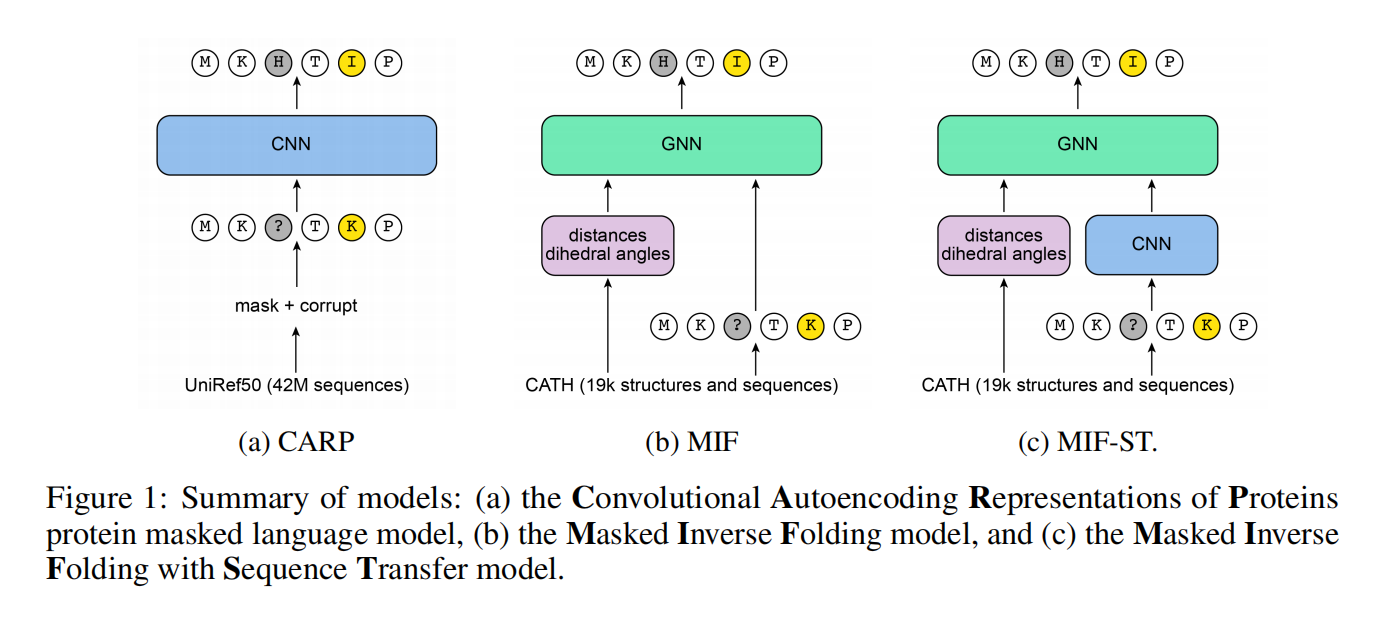
Masked inverse folding
蛋白质的骨架结构由每个氨基酸残基的C、Cα、Cβ和N原子的坐标组成,省去了有关侧链的信息(这将很容易揭示每个残基的氨基酸身份
预训练完了后MIF模型可以直接像sequence-only的PMLM一样直接作用在下游任务上,只是需要提供结构
Embedding backbone structure and sequence
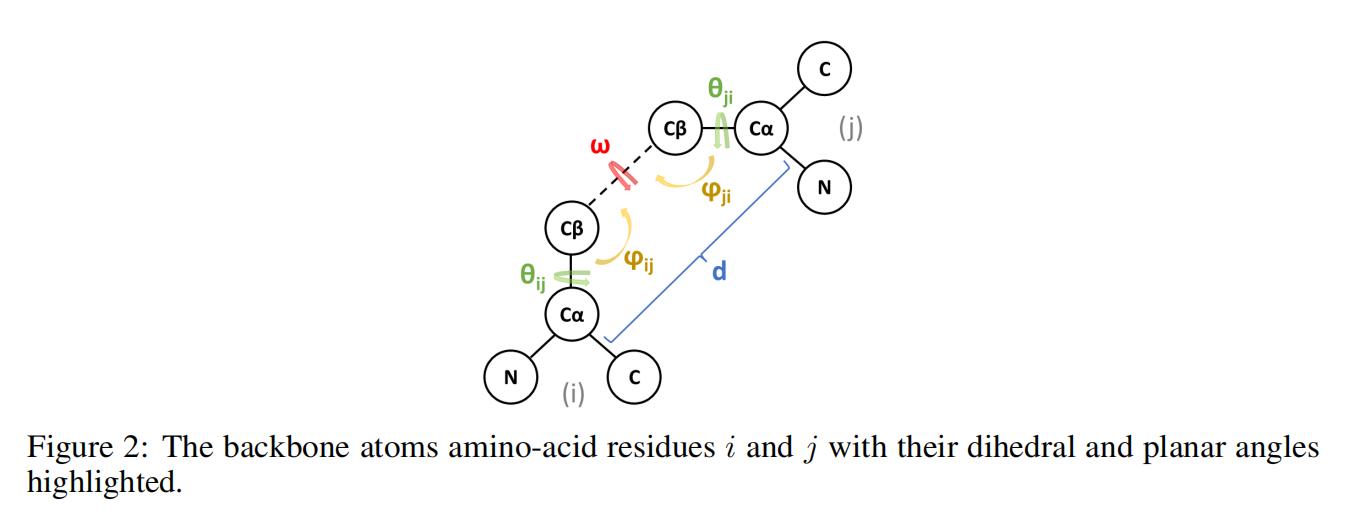
最近邻k=30,每个节点的结构输入特征包括其与主结构中最近邻的二面体角和平面角的正弦变换和余弦变换:
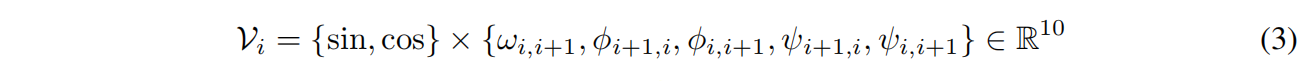
其中 $w$ 是对称的,剩下两个坐标是不对称的,与残基顺序有关,所以两个方向都要编码
输入边特征:
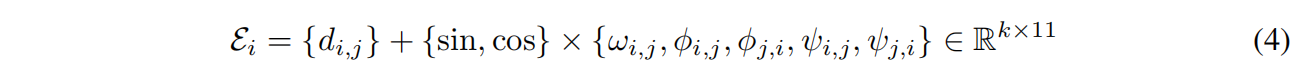
d是残基i和最近邻的Cα之间的欧氏距离
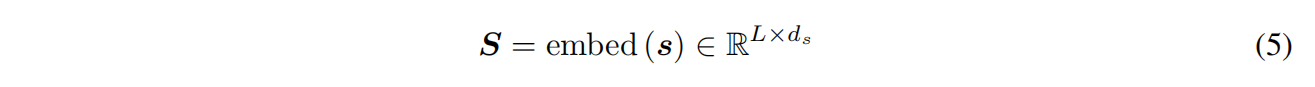
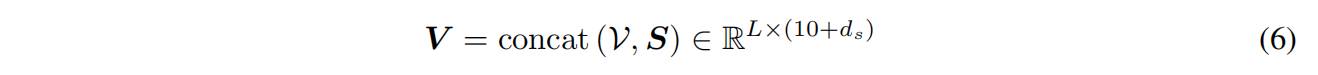
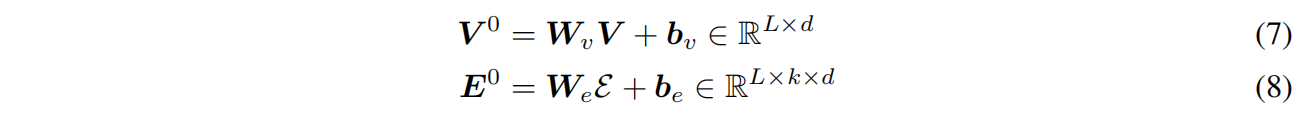
Structured GNN
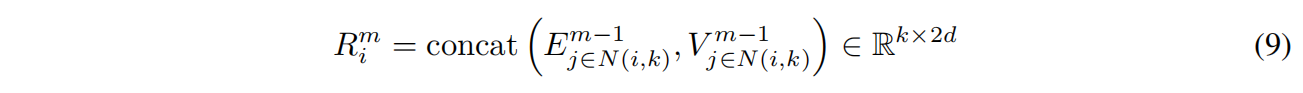
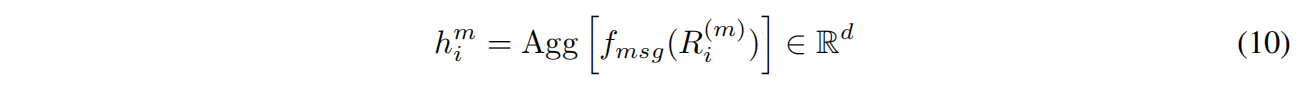
$f_{msg}$ 是三层d维神经网络和ReLU,Agg是平均
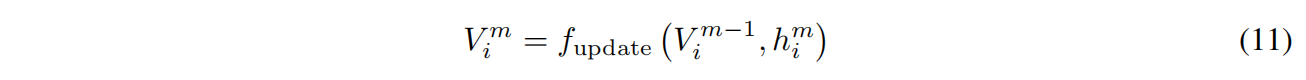
$f_{update}$ 是线性投影
Datasets and model training
CATH4.2数据集,dynamic batch size最大6000个token或100个sequence,Adam优化器,最大学习率1e-3,linear warmup 1000 steps
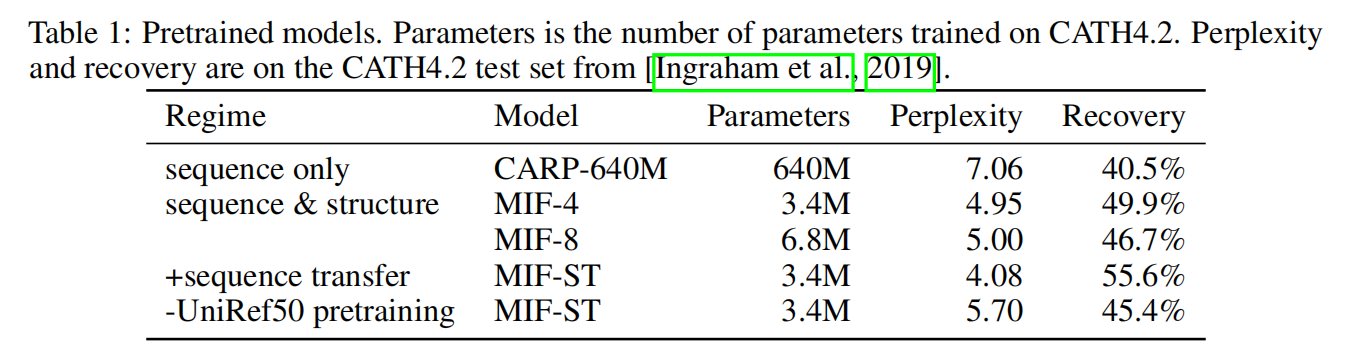
即使MIF比CARP模型参数小很多,但考虑backbone structure情况下还是提高了perplexity和recovery
Masked Inverse Folding with Sequence Transfer
把CARP-640M在UniRef50预训练输出结果替换公式5中的sequence embedding
Downstream tasks
Zero-shot mutation effect prediction
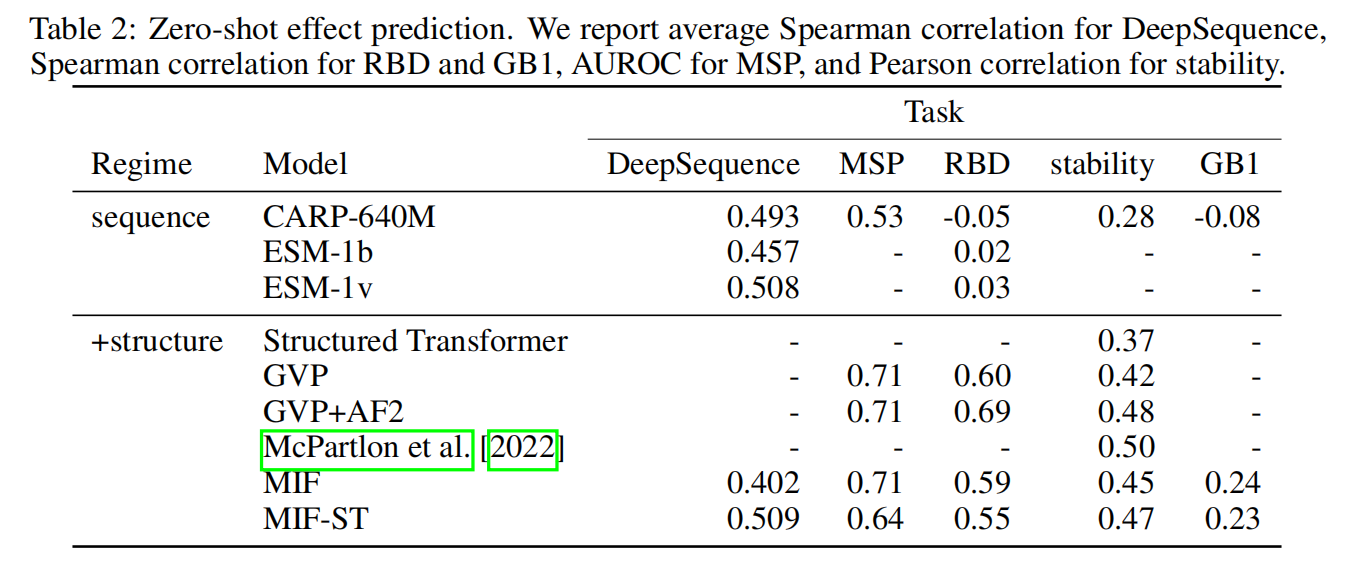
表2都是突变数据集,数据集具体介绍看原文
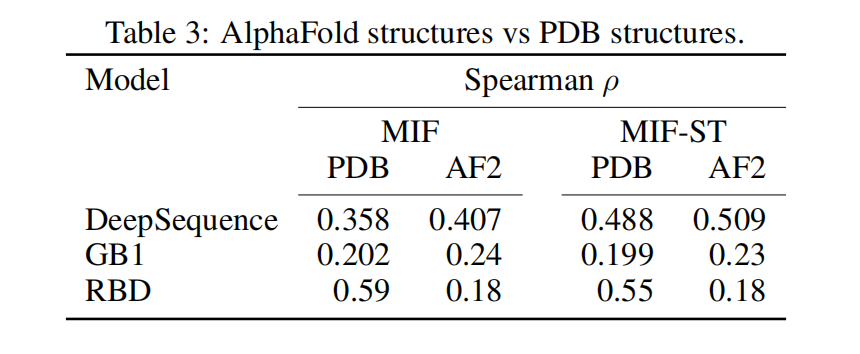
Out-of-domain generalization

在以下两个fitness lanscape进行微调和评估:
- Rma NOD:探索海洋红热霉(Rma)一氧化氮双加氧酶(NOD)酶七个位置的突变如何影响苯基二甲基硅烷与2-二氮丙酸乙酯反应的对映选择性,对214个在3个位置突变的变体进行训练,并将所有7个位置突变的变体随机分成40个验证变体和312个测试变体
- GB1:使用FLIP在上面同样的GB1 landscape分割

pretrained表示是否在Rma NOD上进行了预训练,MIF-ST是唯一一个性能提升的
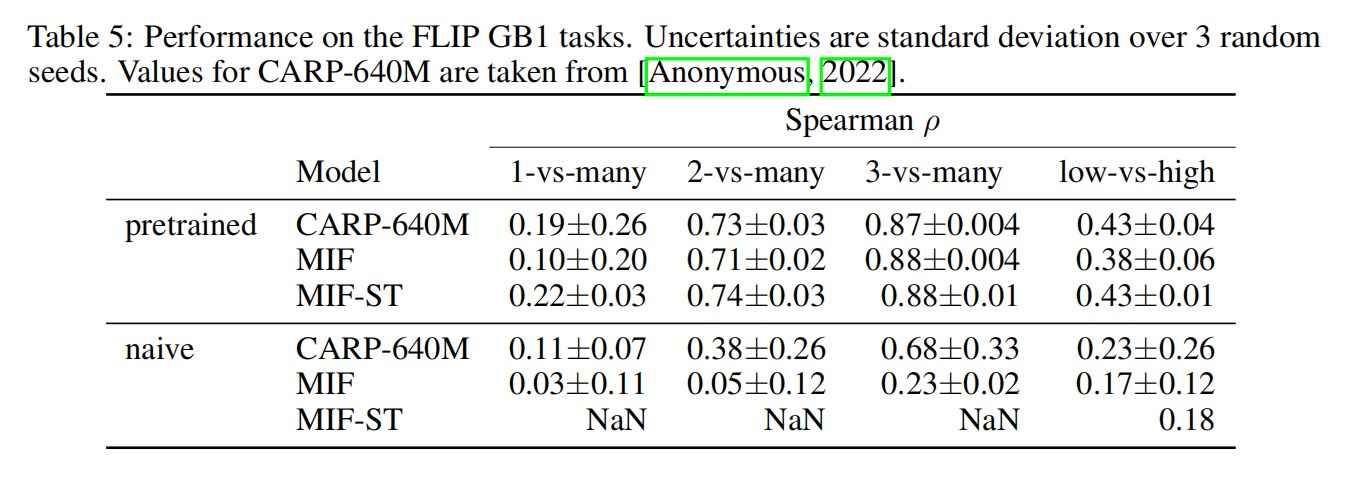
所有的任务都在GB1上预训练得到了提升
Conclusions
Limitations
本项工作对蛋白质和他的变体只使用了一条结构,可以通过预测所有变体的结构来提升结果,但这计算量太大了
此外,因为结构在预训练的适合是保持不变的,还不清楚怎么处理下游任务有插入和删除的情况
Future work
使用更先进的GVP或SE(3)-transformer架构替代Structured GNN
通过AlphaFold结构做数据增强或在输入的结构中添加噪声
使用自回归或span-masking loss来做生成任务,更好的处理插入和删除的情况,更泛化

