论文地址:Tranception:protein fitness prediction with autoregressive transformers and inference-time retrieval
Tranception:卷积多组attention+MSA retrieval
Abstract
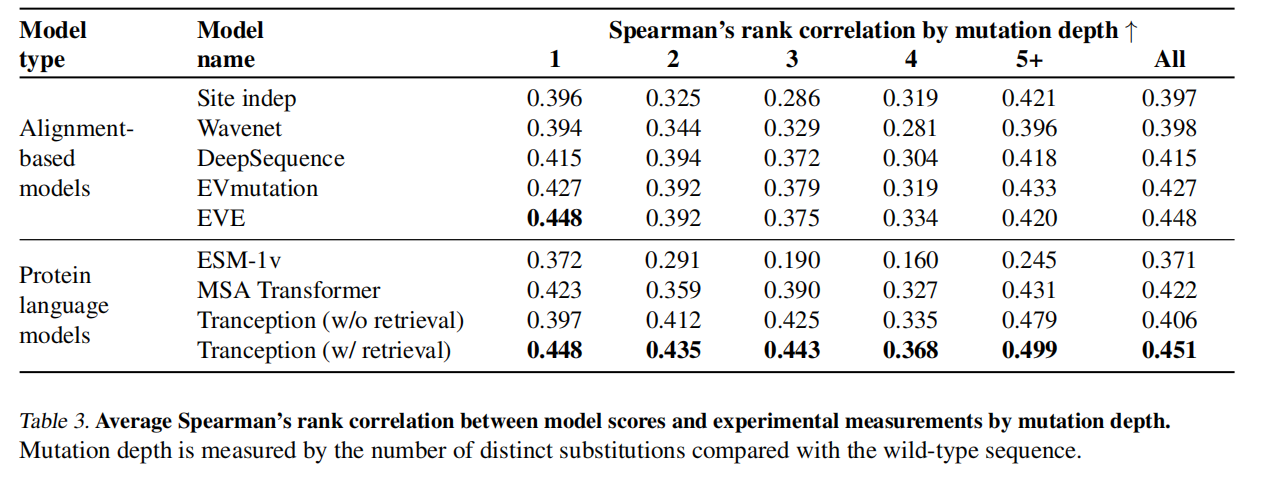
在多序列对齐上训练的深度蛋白质生成模型是目前处理蛋白质fitness最成功的方法,但这些方法的表现取决于是否有足够多和深的alignments来训练,这就限制了蛋白质的家族,大规模语言模型就不需要。提出了tranception,是transformer架构,利用了autoregressive prediction和homologous sequences of inference实现了SOTA。同时提出了ProteinGym数据集
Introduction
目前SOTA方法主要用了MSA,主要两个目的:
- 作为一个数据采集工具,在一个大型的蛋白质数据库中识别与目标相关的序列,然后在一组相关的序列上训练一个模型
- 他们通过建模插入、删除和替换来对齐序列,从而形成一个坐标系统,使给定位置上的氨基酸能够在整个训练集上进行比较
局限性:
- 模型不能对与训练中使用的MSA坐标系不兼容的序列进行预测(例如,插入和删除),从而限制了范围
- 蛋白质组很大部分区域对应的是不能对齐的
- 即使比对是可获得的,蛋白质功能可能是分类特异性的,MSA算法可能无法检索到足够大的同源序列集进行模型训练
- alignment-based模型对训练的MSA比较敏感
- 单独训练在不同数据子集的模型缺少信息共享
基于MLM的模型不能评估整个序列的likelihood导致了预测突变影响时比较启发式,特别是多点位的时候,而且没法打分indels
主要贡献:
- 提出了tranception,transformer架构
- 结合了autoregressive prediction和homologous sequences of inference达到SOTA
- 提出了proteingym数据集
Tranception
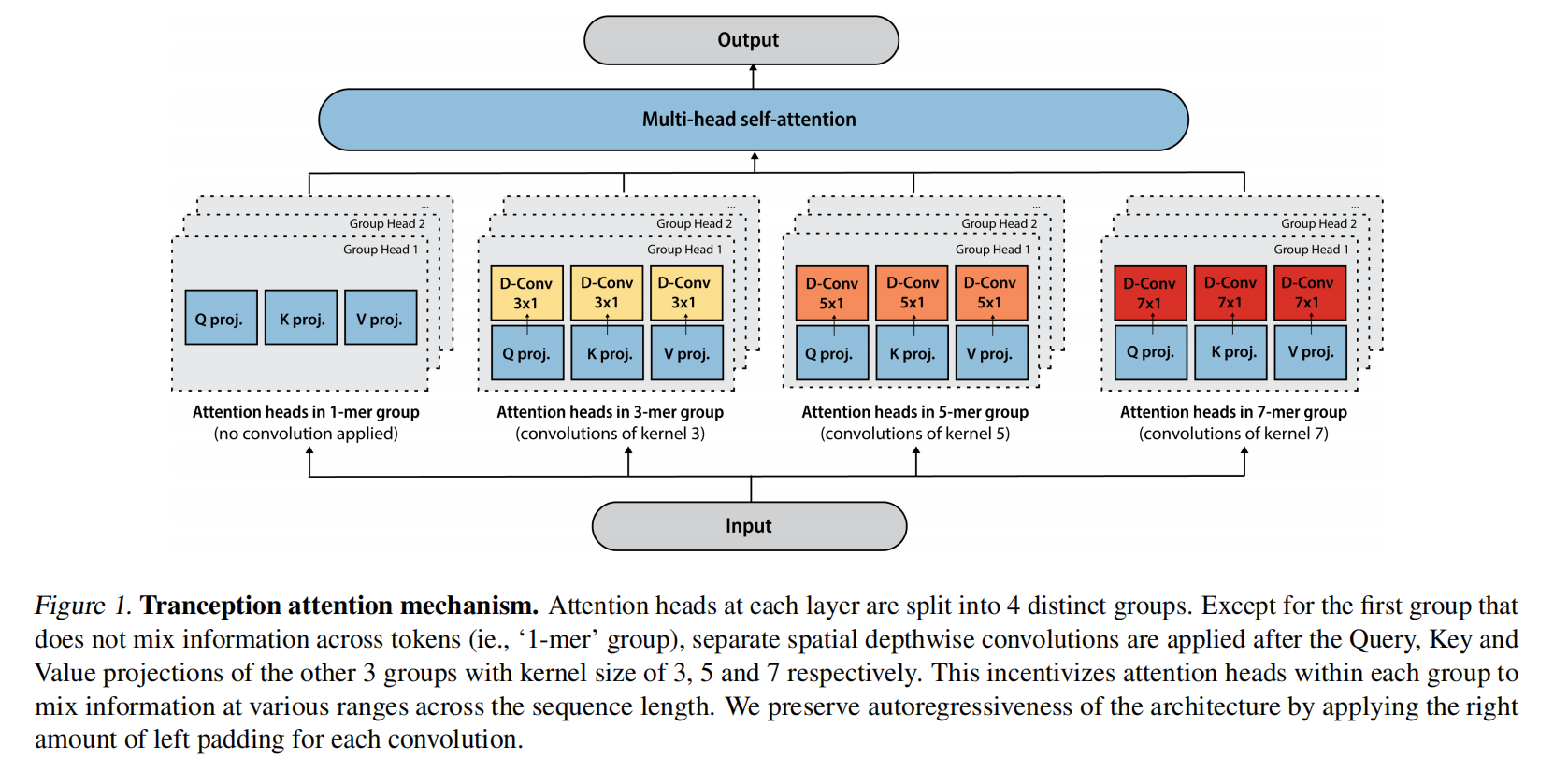
Tranception attention
研究了新的注意力机制,来关注氨基酸token的连续子序列(k-mer),把每一层得注意力头分为4组,每一组应用不同核大小的卷积
Grouped ALiBi position encoding
把可学习的位置编码或是sin位置编码换成了ALiBi的一个变体,称为grouped ALiBi
Data processing and augmentations
模型参数700M,训练集是UniRed100,有250M的序列
最大序列1024,如果训练时蛋白质超过1024就随机选择连续的片段达到这个最大长度
Scoring sequences for fitness prediction
训练目标是自监督的,给定i-1个token预测第i个token
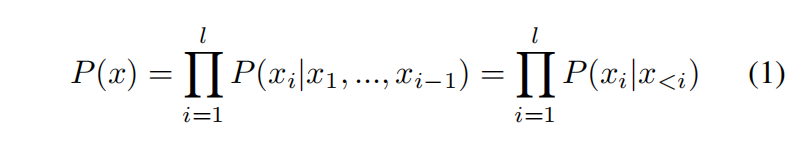
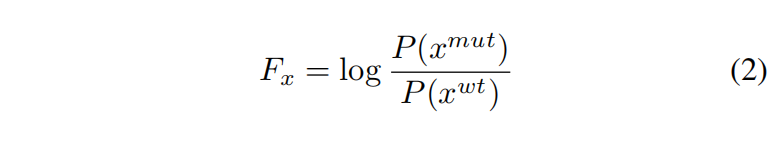
对于超出最大长度限制的蛋白质,选择可以提供最多突变的序列片段
Inference-time retrieval
Multiple sequence alignments
在一个给定的位置上,观察到的MSA序列上的氨基酸分布概括了进化约束:属于MSA的蛋白质序列是保持适应度的变体,并且没有被进化选择淘汰
Two modes of inference
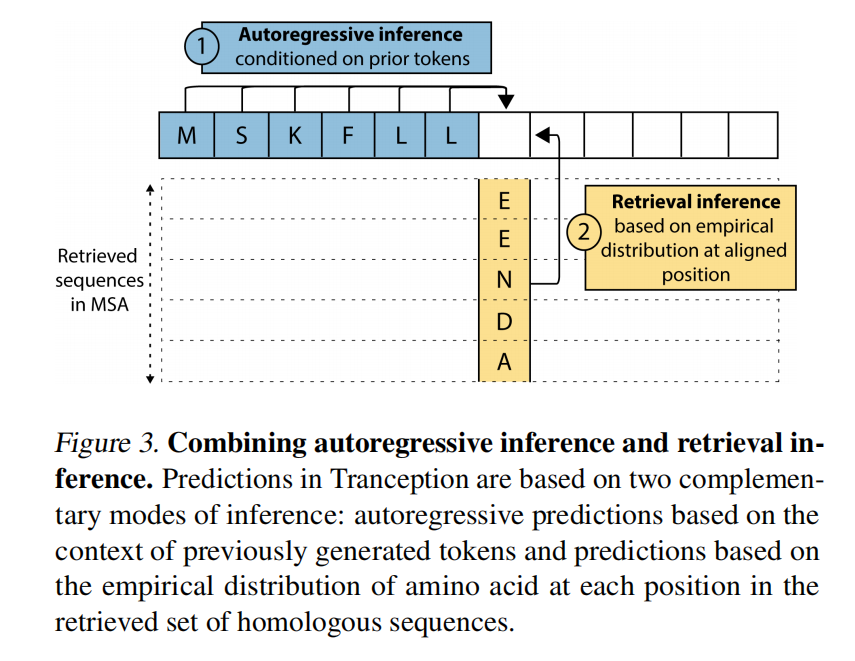
第二种模式:retrieval inference
第一步在推理阶段要限制检索的MSA:
- 替换:检索到的同源序列集对于野生型和突变序列都是共同的:我们每个家族进行一次检索步骤,并对所有要评分的突变序列分摊成本
- 插入和删除:我们通过删除MSA中与删除位置相对应的列,并在MSA中的突变蛋白插入位置添加零填充列,将检索到的MSA调整为每个突变序列
第二步:
logP_A是自回归的概率,logP_R是检索推理得到的概率,C是正则化常量
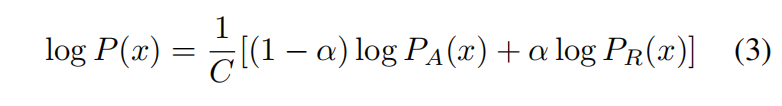
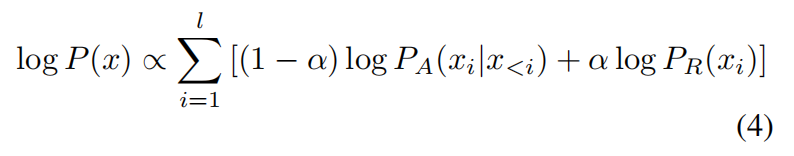
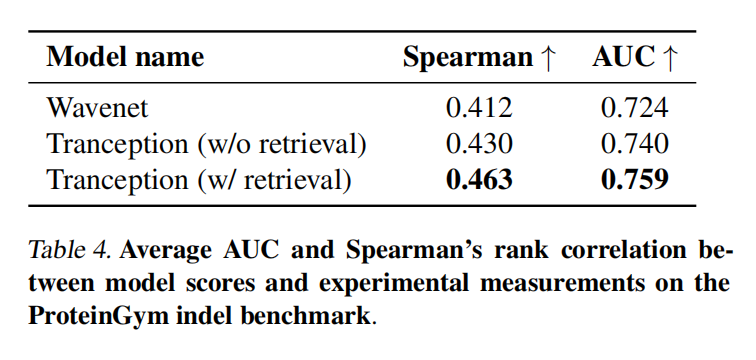
Results