论文地址:Accurate and efficient protein sequence design through learning concise local environment of residues
ProDESIGN-LE:transformer编码局部结构设计蛋白质序列
Abstract
提出ProDESIGN-LE用于高效的做蛋白质序列设计,采用了一种简洁但语义丰富的残基局部环境表征,并训练了一个transformer来学习残基局部环境和氨基酸type之间的相关性。对于一个给定的backbone structure,ProDESIGN-LE通过transformer根据该结构中的局部环境为每个位置分配合适的残基类型,最终获得所有残基与局部环境匹配良好的设计序列。应用ProDESIGN-LE可以为平均每个蛋白在20秒内设计68个自然发生和129个幻觉蛋白序列,预测结构与目标结构完全相似,最好的TM-score有0.8。
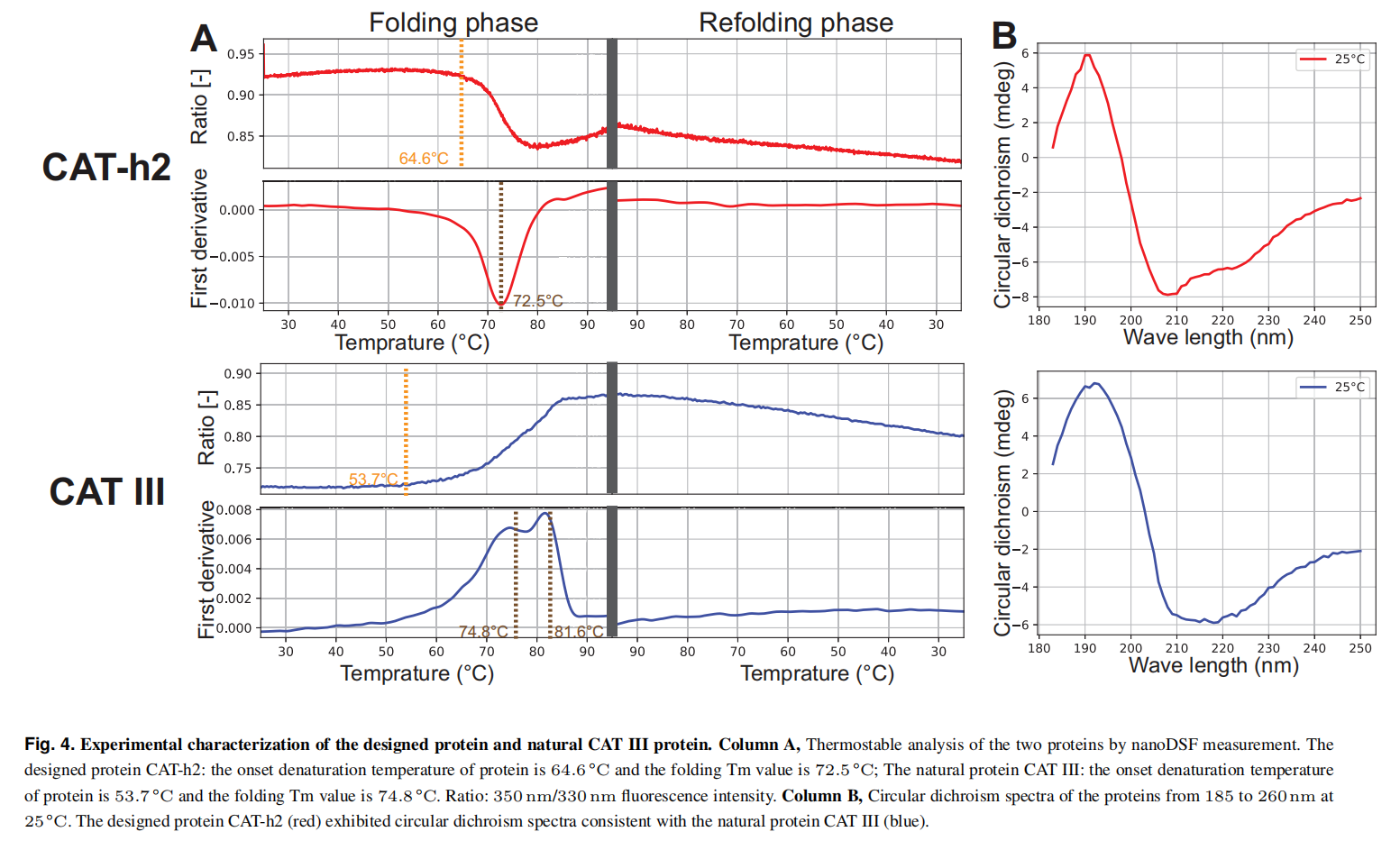
通过设计氯霉素o-乙酰转移酶III型(IITIII),并在大肠杆菌中重组表达,进一步实验验证了ProDESIGN-LE。在这些蛋白中,有三种具有良好的溶解度,其中一种产生了与天然CAT III蛋白一致的圆二色光谱单体
Introduction
蛋白质序列设计可以使用同时发生策略实现,即同时确定所有蛋白质残基的氨基酸类型,大多数这种类型的方法都寻求利用来自目标主干结构的残基间距离。比如SPROF把应用CNN从残基间距离map中推理出蛋白质序列,或是ProteinSolver用深度图网络来发现满足残基间约束的蛋白质序列,但这些方法泛化能力差
蛋白质序列也可以迭代设计,每次迭代随即算一个残基突变来观察是否提升了fitness,比如用Rosetta打分
因为残基-残基之间的交互的局部性质,大多方法都是利用目标结构中特定残基的局部环境,比如几何特征(周围残基的相对距离),理化性质(邻近氨基酸类型和溶剂可及性solvent accessibility)来预测目标残基
我们的方法的基本原理是,一个设计的蛋白质,如果每个组成的残基都符合目标结构定义的局部环境,就可以折叠成一个类似于目标结构的全局结构
Approach
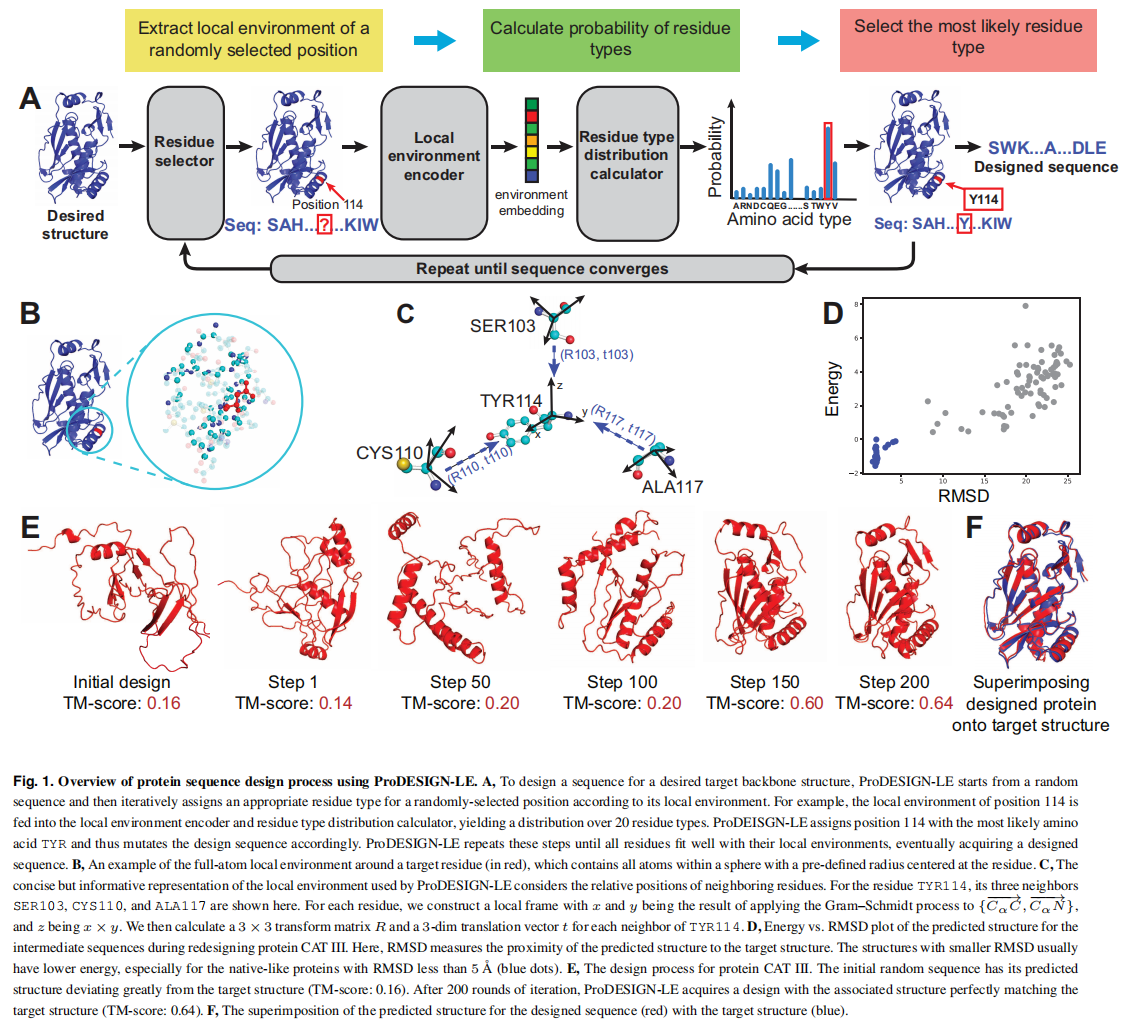
每个氨基酸的坐标可以表示为 $B_i=(N-i,C_{\alpha_i},C_i)$
局部环境表示为 $env_i={(s_j,B_j \ominus B_i,j-i)|\parallel C_{\alpha_i}-C_{\alpha_j} \parallel \le T }$,$\ominus$ 符号表示计算相对距离,给定骨架B设计序列S可以表示为
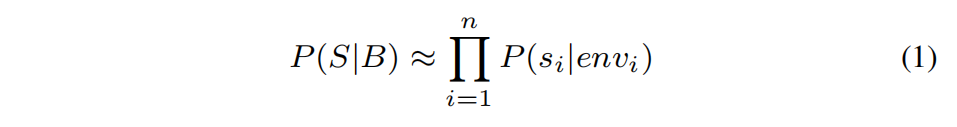
也就是最大化这个概率
算法流程,从一个随机序列开始然后重复执行以下步骤:
- 随机选择位置i
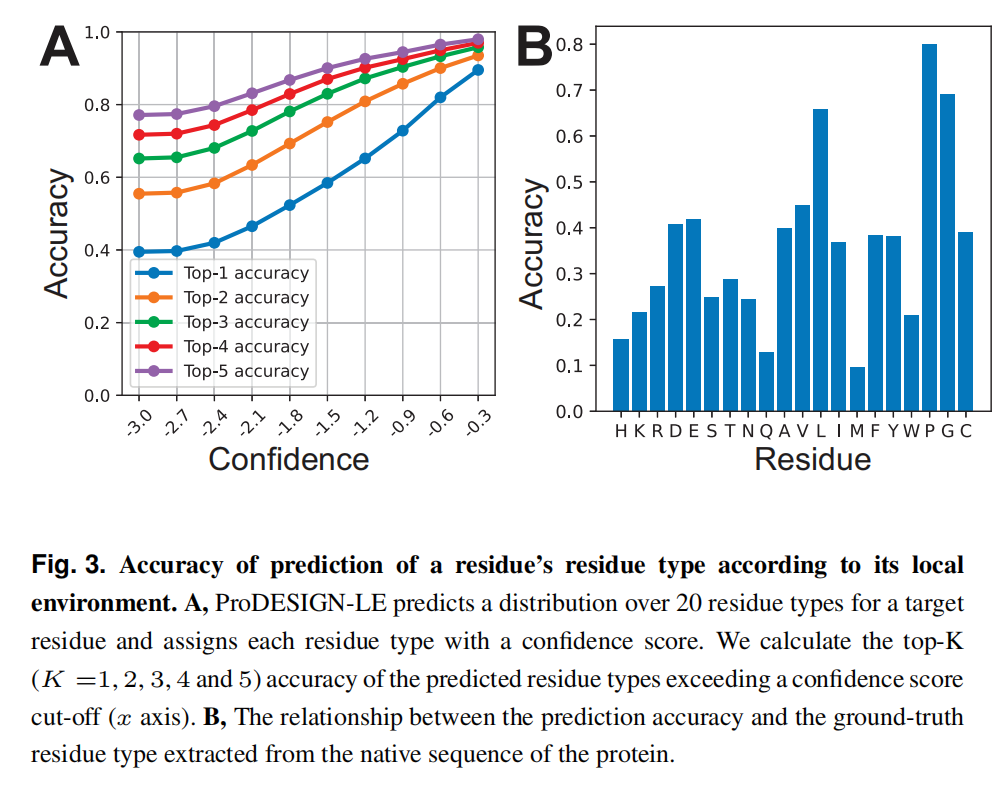
- 对每个可能的氨基酸a的type计算条件概率 $P(a|env_i)$
- 突变 $s_i$ 为最可能的氨基酸type $s_i\leftarrow argmax_aP(a|env_i)$,同时局部环境信息也更新
使用transformer做分类器,PDB40数据集的子集训练,对于局部几何环境 $env_i$ 有两个氨基酸的相对位置信息表示为3x3的旋转矩阵和3x1的平移向量,flat之后和其他信息比如(one-hot编码,相对序列位置)concat起来
Materials and methods
Network architecture and training procedure
(1)Local environment encoder:三层transformer,每层16个头
(2)Residue type distribution calculator:全连接层+softmax
batch_size=1000,Adam优化器($\beta_1=0.9,\beta_2=0.999$),lr=1e-3
Datasets
PDB40
从9,995个蛋白质结构中提取的5,867,488个残基的训练集和一个包含从401个蛋白质结构中提取的758,160个残基的测试集
局部特征包含:
- 相邻氨基酸的one-hot编码
- 3D相对位置
- 目标氨基酸与相邻氨基酸的距离
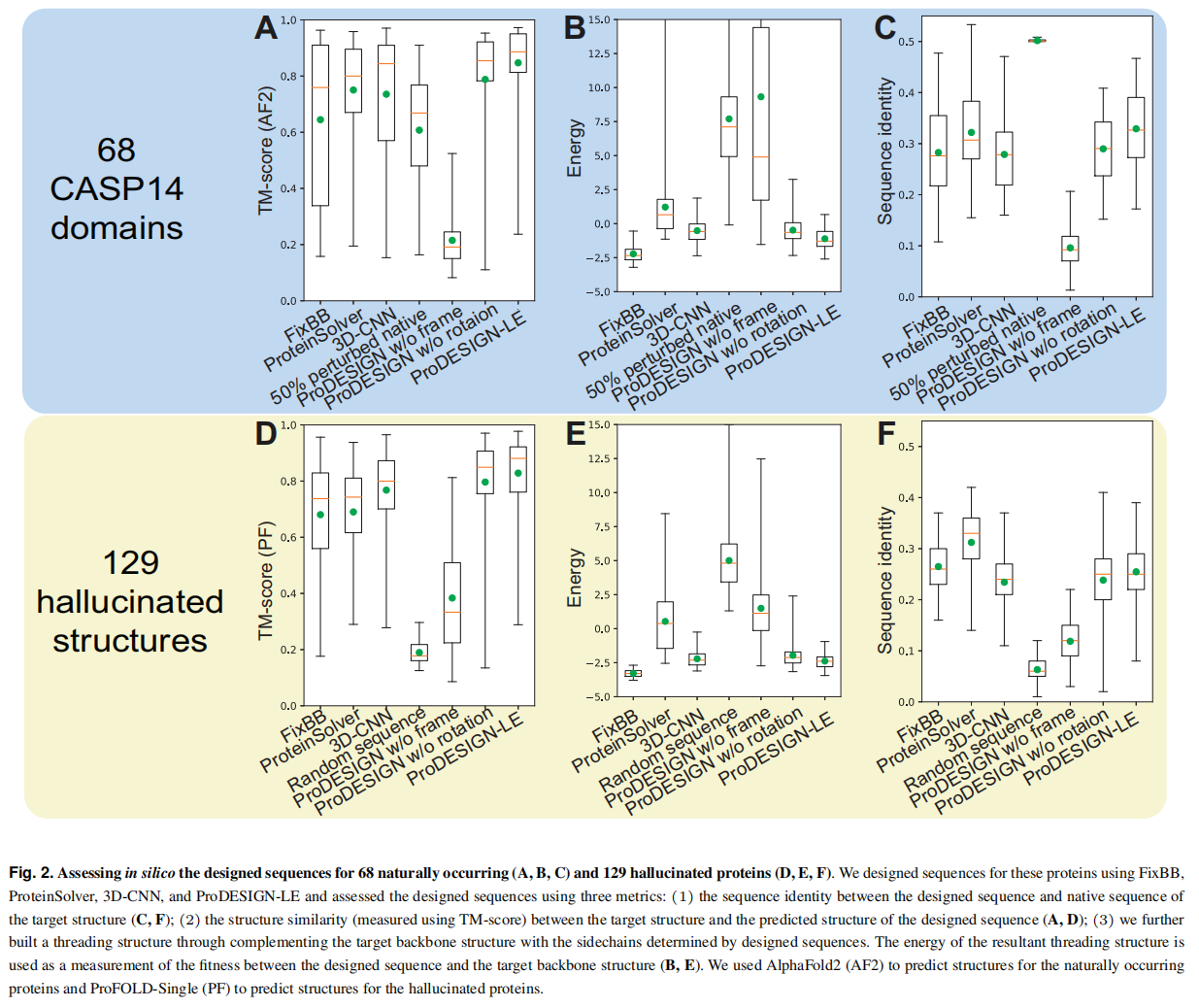
使用从CASP14中提取的68个天然蛋白和129个幻觉结构,测试了ProDESING-LE在序列设计中的性能。为了避免重叠,过滤掉了与这68个CASP14蛋白相似的训练蛋白(相似性截止:e值<1×10−3)
Predicting 3D structure for the designed proteins
利用所设计的蛋白质的预测结构和目标结构之间的接近性来评估一个所设计的蛋白质,用alphafold2和作者的ProFOLD-Single
Protein expression and purification
…
Analyzing protein thermal stability
…
Circular dichroism spectroscopy
…
Analytical ultracentrifugation
…
Results and discussion