论文地址:Learning protein fitness models from evolutionary and assay-labeled data
论文实现:https://github.com/chloechsu/combining-evolutionary-and-assay-labelled-data
CEALD:突变氨基酸one-hot编码+fitness有监督训练
Abstract
基于机器学习的方法预测protein fitness要么从无标注的进化相关的序列中学习,要么从实验验证的有标签的变体序列中学习。作者提出一个简单的组合方法能够超过其他更精致的模型。主要是在site-specific amino acid features上使用岭回归,结合一个从进化序列中建模出的概率密度特征。发现使用变分自编码器的probability density model能取得最好的效果
Introduction
实际中我们需要对蛋白质进行reengineer来使得其更好符合我们需要的功能,比如提高酶活性,使荧光蛋白更亮,修改原本的功能到一个相关到不同的功能等,这里我们主要关注使用机器学习来建模protein fitness prediction
有两种主流的基于机器学习的策略来评估protein fitness
- 使用隐式fitness约束,自然界有的蛋白质序列即进化数据,搜索同源序列建模估计这组蛋白质序列的概率密度模型
- 虽然这些进化相关的同源序列集没有直接和我们需要评估的蛋白质性质关联,但同源搜索本身提供了一个隐式的,弱的,正面的label约束。相比于纯粹的无监督学习而言,对于learning而言是有weak-positive相关的
- 训练这样的case需要数百到数百万不等
- 使用在实验数据上得到label的变体序列做有监督回归训练
- 有监督的数据在数百到数百万不等
- 突变的序列集合可能被限制在1,2个突变或是分布更不均匀
- 因为这样数据有限,所以其实不能全面描述protein fitness landscape,但是一个很好的起点
最近又一些工作结合了两者,对进化相关的序列进行weak-positive学习,同时对有标注的蛋白质变体进行有监督学习。但是对于许多有实际意义的场景,蛋白质的标注只有数百条,因此结合两种方法是很有必要的
Results
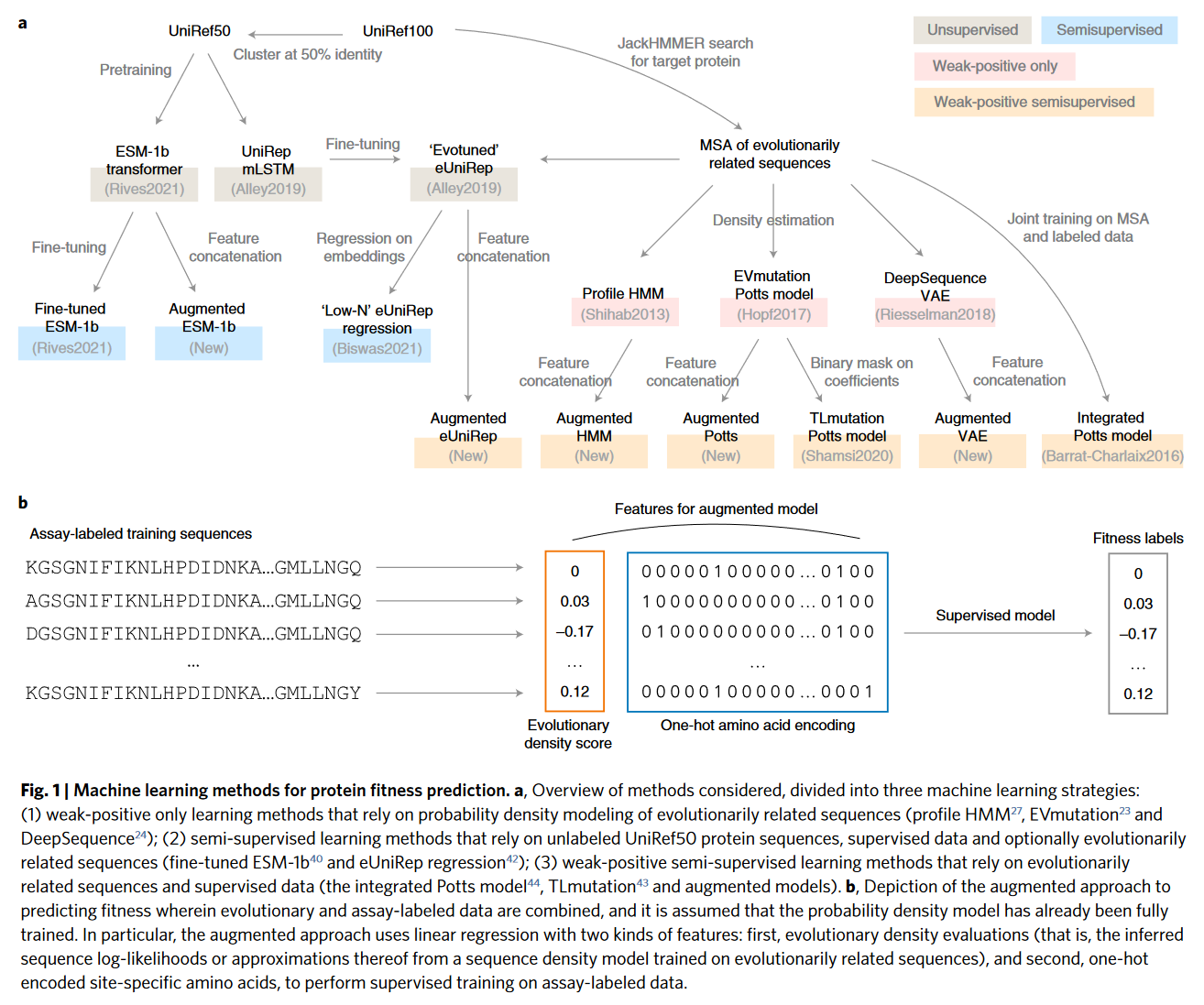
Published machine learning methods assessed
我们评估了13种预测蛋白质fitness的机器学习方法,使用了进化数据,实验数据或都用了,有些还利用了额外的训练数据比如uniref50
三种纯进化模型:hidden Markov model (HMM),Potts model (EVmutation)和VAE (DeepSequence)
两个有监督模型:用实验数据来做One-hot encoded site-specific amino acid fetures的线性回归模型,用esm1b在实验数据集上有监督微调的模型
A simple baseline approach
我们提出了一个简单的方法能够结合进化和实验数据,能够于其他更复杂或昂贵的方法达到competitive
对于任何已经训练过的进化概率密度模型,我们对一个one-hot的特定氨基酸特征进行岭回归,用来增强序列密度评估,把这种方法称为”augment“
因为在回归增强中只使用了位点特异性的氨基酸特征,所以贝叶斯更新主要集中于位点特异性的参数
Assay-labeled and evolutionary data sets
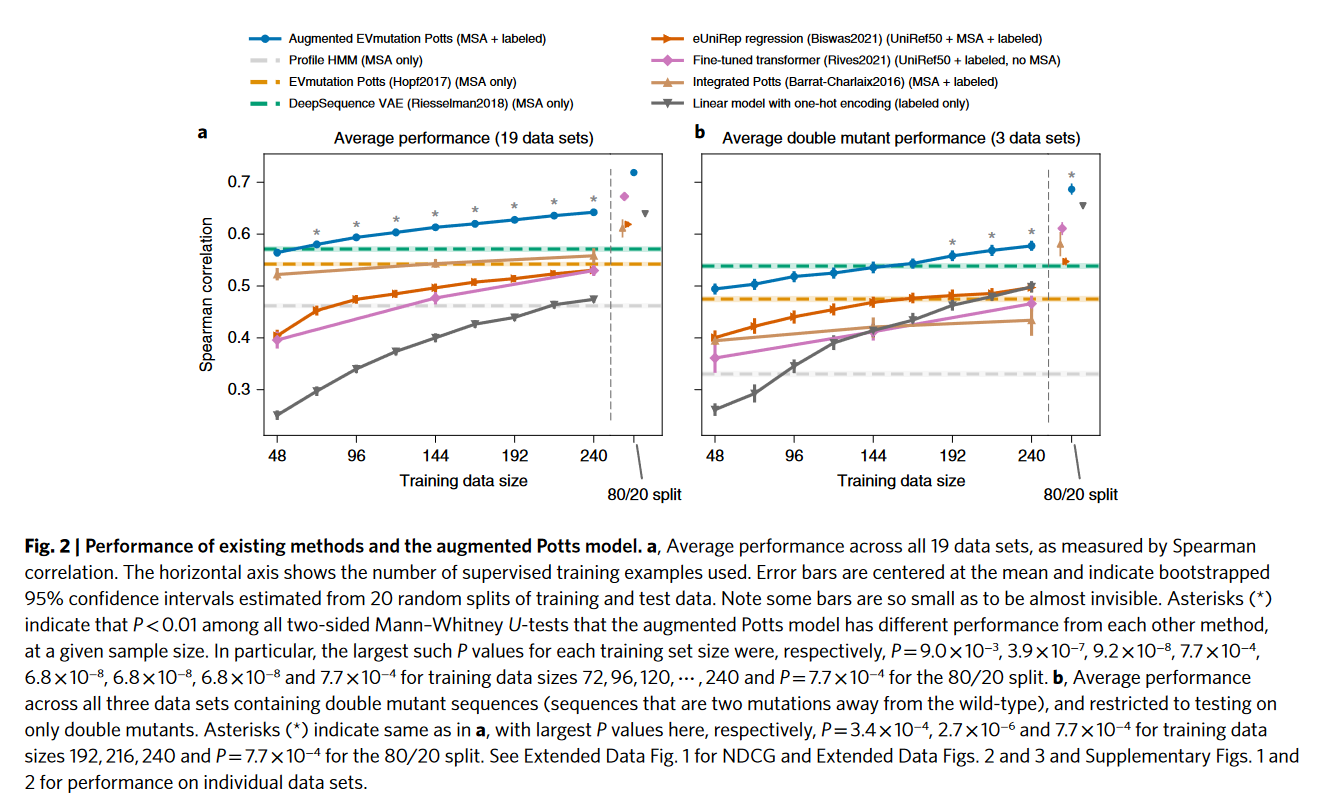
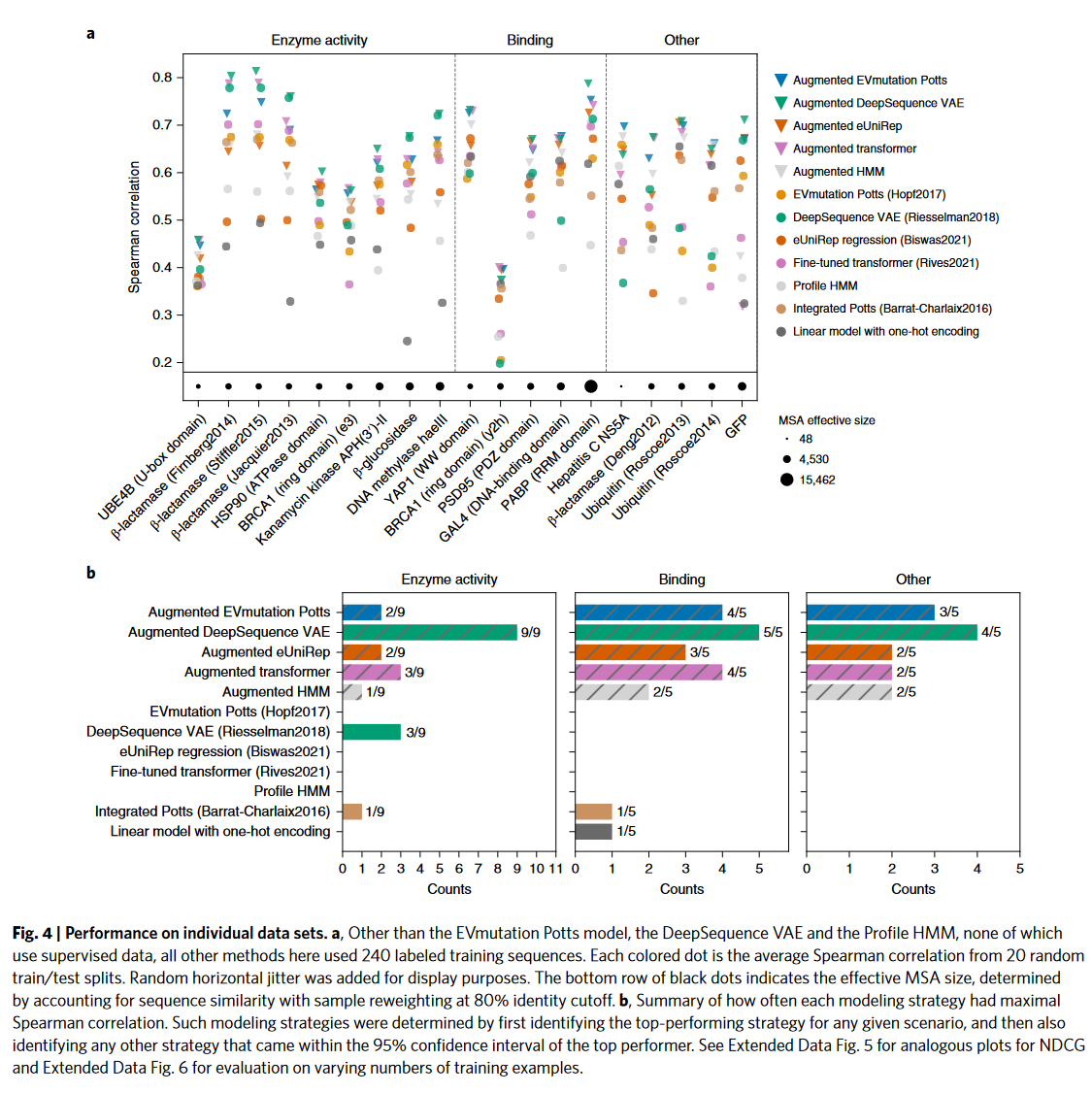
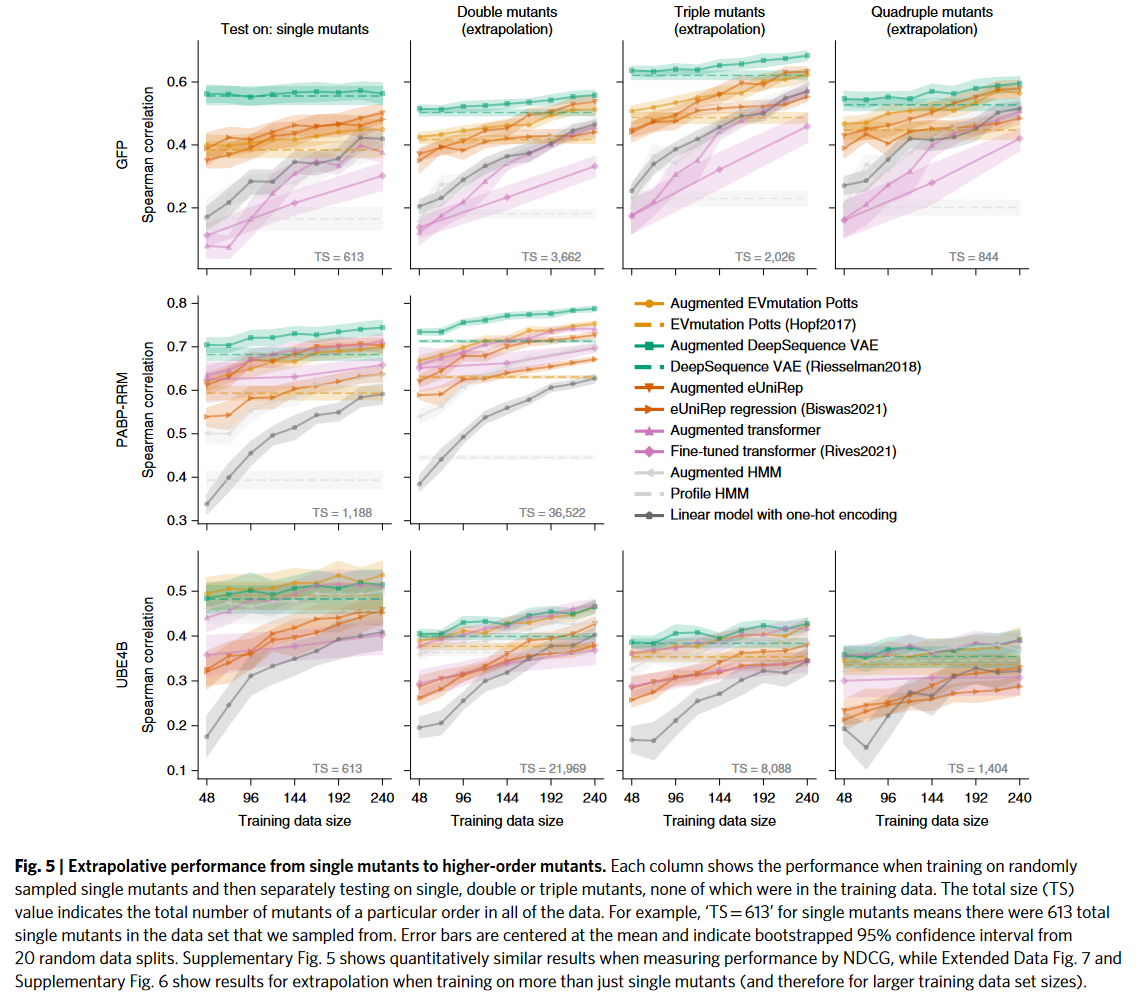
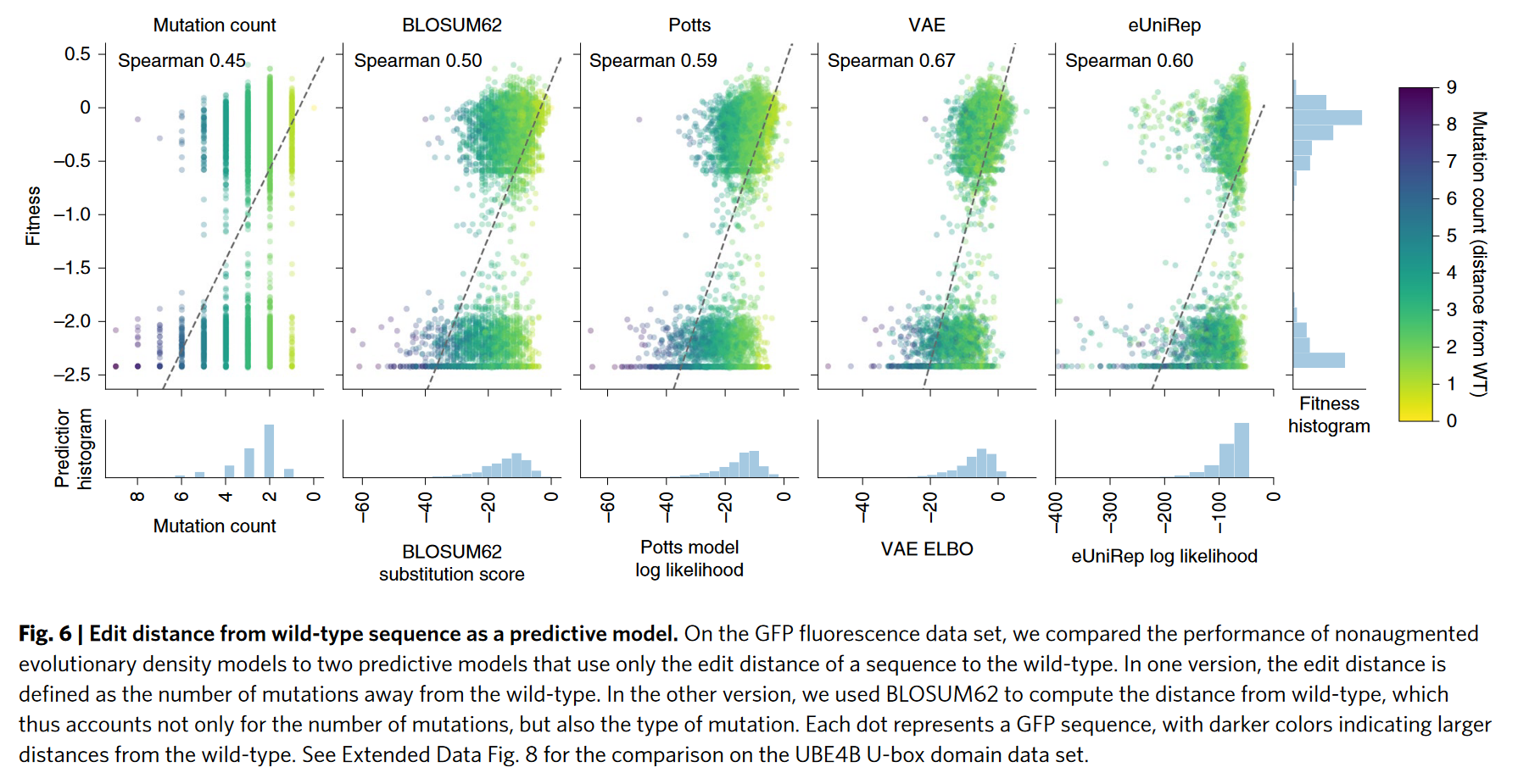
在19个突变数据上进行了评估,包含了EVmutaion的所有数据和GFP的数据集,其中16个只包含单点位突变,MSA是直接使用EVmutation提供的
Experimental overview
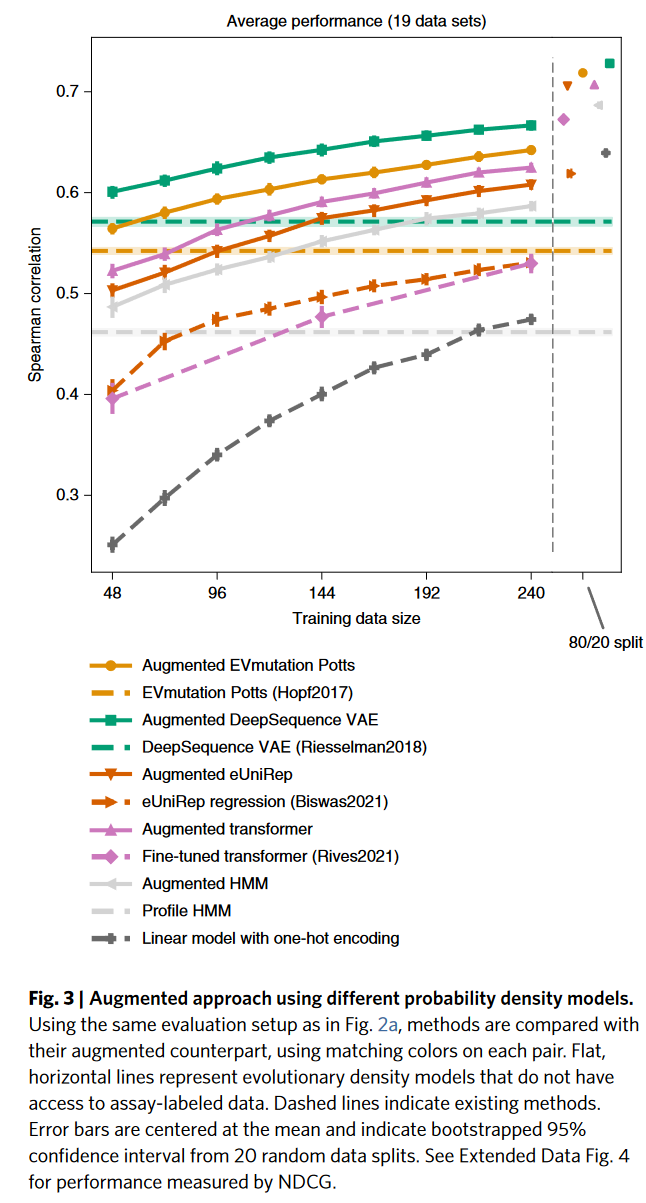
每个数据集用了20%做测试集,然后增加训练样本数
使用了两个指标评估模型性能:spearman rank和normalized discounted cumulative gain (NDCG)