论文地址:FLIP:Benchmark tasks in fitness landscape inference for proteins
FLIP:蛋白质fitness有监督预测benchmark
Abstract
机器学习方法来捕获蛋白质序列-功能的关系,被称为fitness landscape。目前的benchmark像是CASP和CAFA分别是评估蛋白质结构和功能预测,但和蛋白质工程不相关。本文介绍一个fitness landscape inference for proteins(FLIP)的benchmark来让蛋白质工程领域的表征学习进行快速预测。FLIP包括adeno-associated virus stability for gene therapy(腺相关病毒基因治疗稳定性)、protein domain B1 stability和immunoglobulin binding以及多个蛋白家族的热稳定性实验数据
Introduction
蛋白质执行所需功能的能力是由其氨基酸序列决定的,通常是通过折叠到三维结构来介导的
目前的生物物理和结构预测方法不能可靠地将序列映射到其执行所需功能的能力,所以目前蛋白质工程在很大程度上依赖于定向进化(DE)的方法,随机修改(突变)一个起始序列,测量所有序列找出fitness提高的,然后迭代直到蛋白质性能足够。从序列中预测适应度的机器学习方法可以利用正负样本选择筛选突变序列,比传统的定向进化以更少的测量量达到更高的fitness的效果,不需要对结构或机制的详细理解
Related Work
The Critical Assessment of Protein Structure Prediction (CASP) 做蛋白质结构预测
The Critical Assessment of Function Annotation (CAFA) 重点是将基因本体论(GO)类(蛋白质功能的分类定义)分配给蛋白质
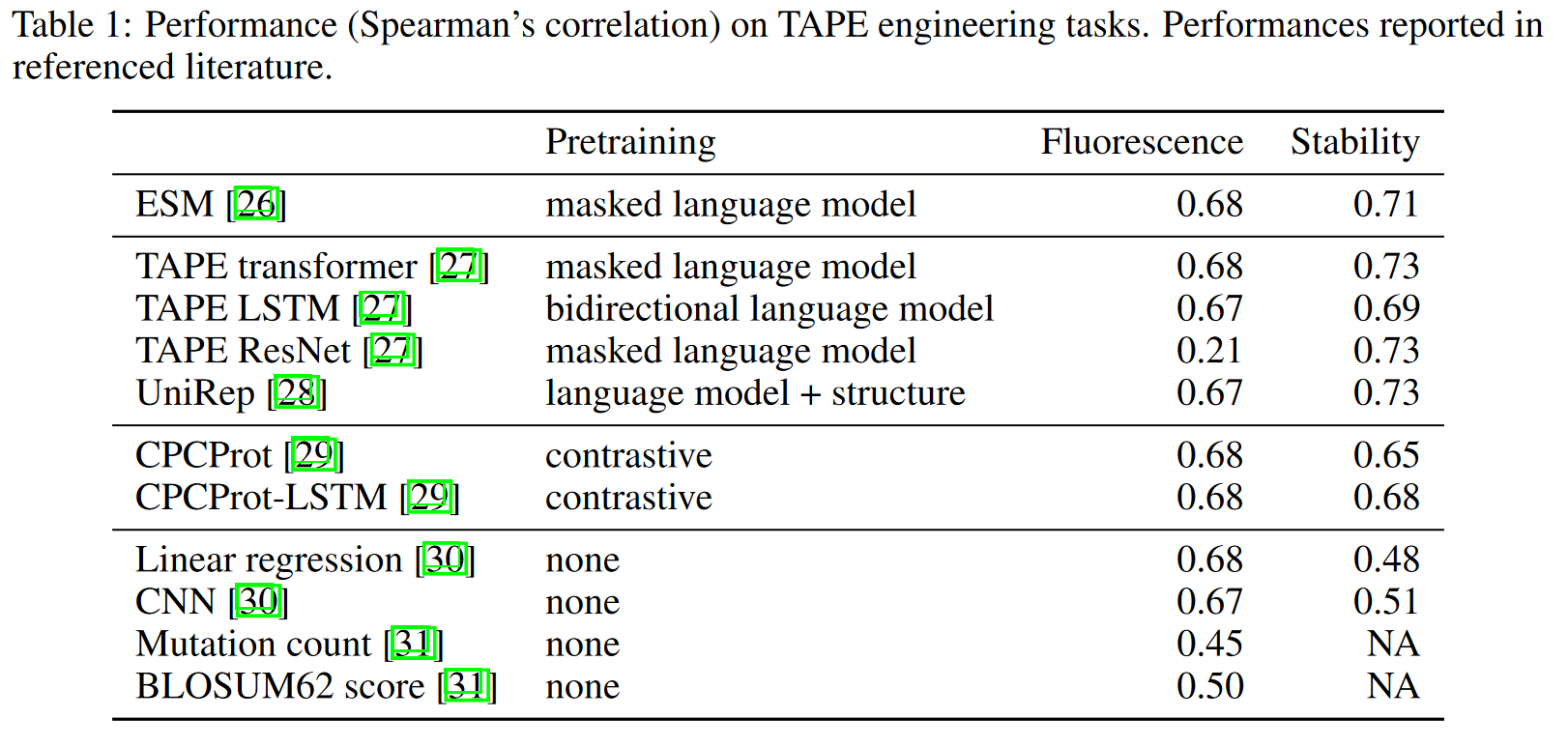
Tasks Assessing Protein Embeddings (TAPE) 评估不同的预训练机制和模型对预测蛋白质特性的有效性
Envision整理了几十个单一氨基酸变异(SAV)数据集,但不包括其他类型的序列变异
DeepSequence收集了42个深度突变扫描(DMS)数据集
这些研究没有一个标准的train/test split
Landscapes and Splits
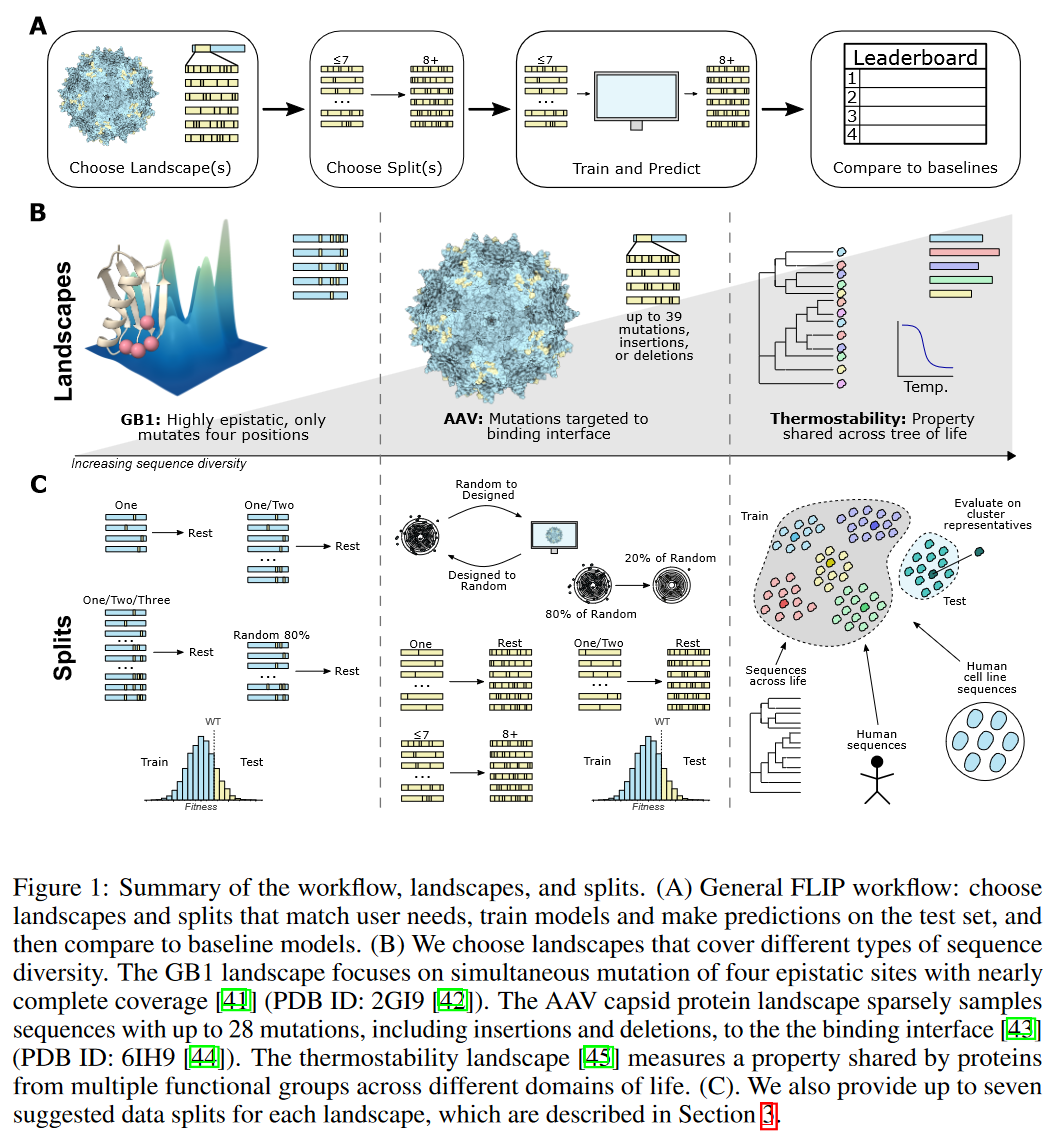
设计FLIP来回答两个关于机器学习建模蛋白质序列的基础问题:
- 一个模型是否能捕捉到除了父序列突变之外的复杂fitness
- 一个模型能否在一系列测量非常不同功能的适应度的蛋白质上表现良好
现有的工作如DeepSequence、Envision等可以很好的证明第二点,但不能证明第一点
TAPE的荧光预测任务能评估第一点,但不能评估第二点
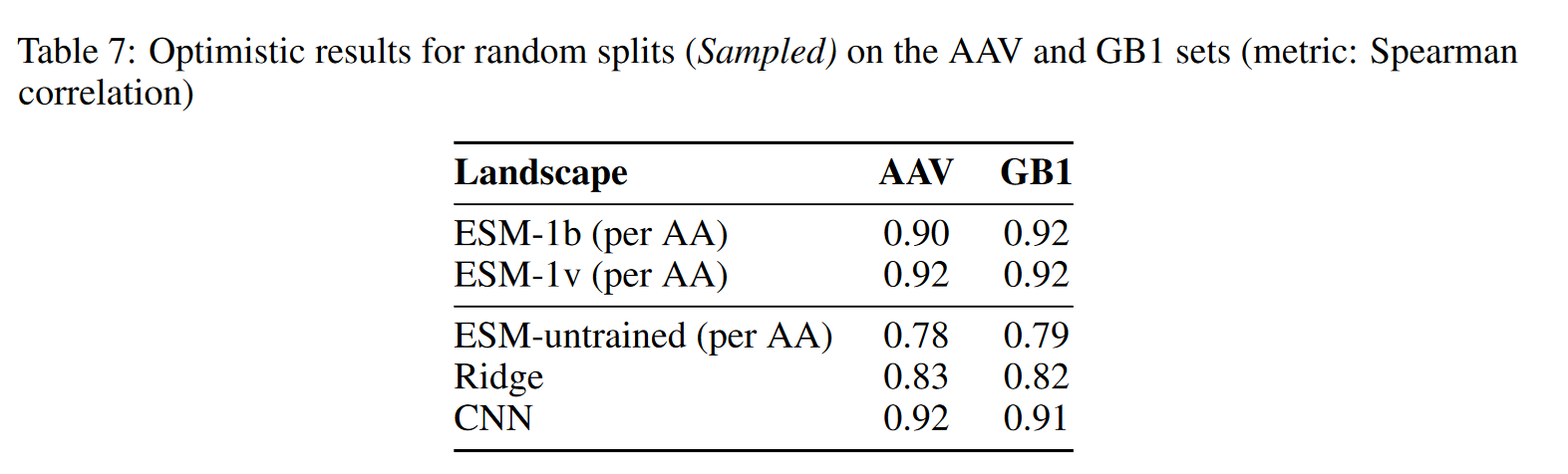
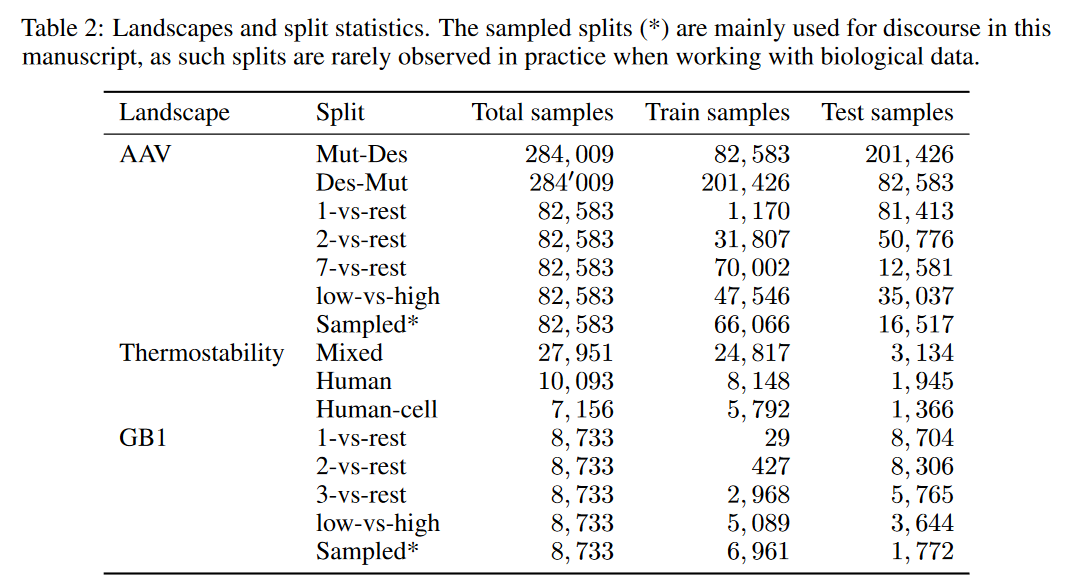
为了测试上述问题,在表2收集了三个已发表的landscape并创建了15个split,来测试模型不同情况下的泛化能力。简单的随机分割方法在经典的蛋白质序列到功能的预测中是出了名的误导性,因为蛋白质序列不是采样的I.I.D,而是与进化历史是相关的
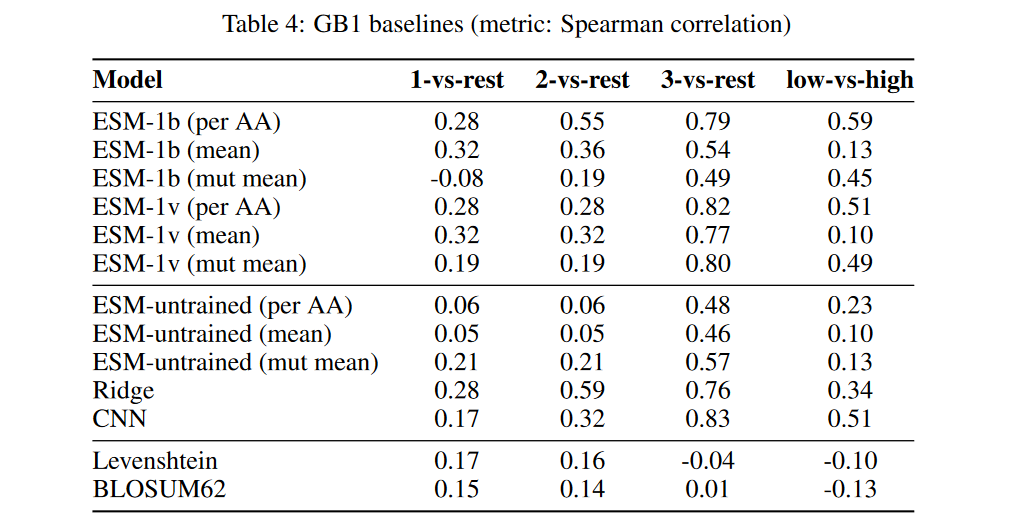
GB1
Motivation
预测突变之间的相互作用的能力称为上位性,这些相互作用导致了对蛋白质适应度的non-additive效应,并已被证明限制了进化可用的路径
Landscape
GB1是蛋白G的结合域,蛋白G是链球菌中发现的一种免疫球蛋白结合蛋白。在他们最初的研究中测量了在4个位置的16万种可能的突变组合中的149,361种的fitness
Splits
超过96%的氨基酸突变产生非结合或低结合序列149361个序列中有143539个的适应度值低于0.5,其中野生型fitness为1,fitness为0为非结合
为了确保模型学习nontrivial信号,在创建训练集之前对非功能序列进行降采样。5822个适应度以上的0.5的序列和2911个适应度小于或等于0.5的随机抽样序列。从这个集合中,策划了5个数据集的分割
- Train on single mutants (1-vs-rest)
- Train on single and double mutants (2-vs-rest)
- Train on single, double and triple mutants (3-vs-rest)
- Train on low fifitness, test on high (low-vs-high):负样本训练,正样本测试
- Sampled:随机划分8:2
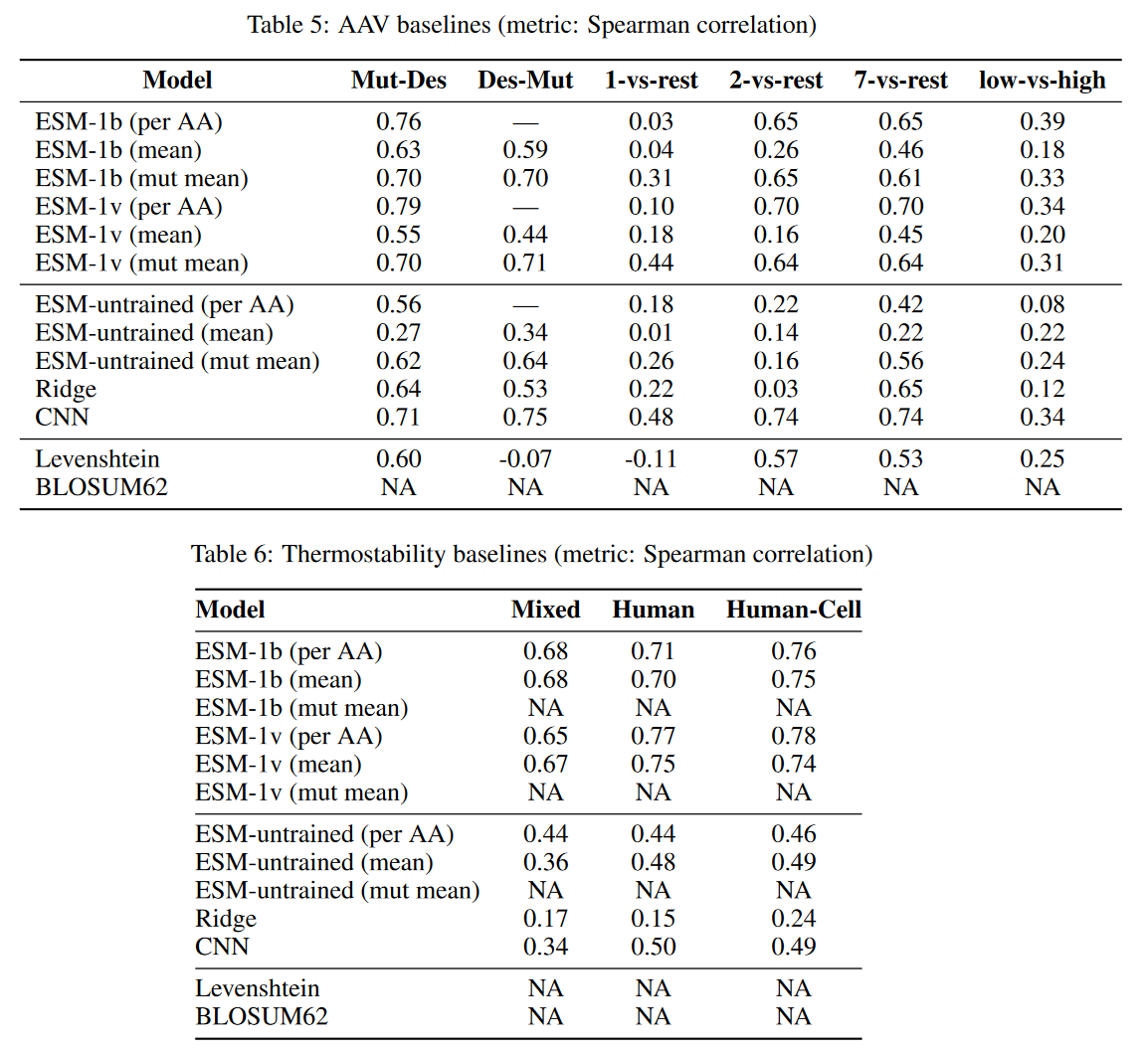
AAV
Motivation
蛋白质工程上往往只突变一个特定区域,比如已知protein-protein interface是知道作为subset of positions,对长序列而言成功预测这样一个位置子集也是有意义的
Landscape
Adeno-associated virus (AAV) 衣壳蛋白负责帮助病毒将DNA有效载荷整合到目标细胞中。
从VP-1的561到588位有一个28个氨基酸窗口,测量1到39之间的变异的适应度,这被称为采样池(sampled pool)。此外,还测量了使用各种机器学习模型选择或设计的序列的适应度,这些称为设计池(designed pool)
Splits
- Sampled-designed (Mut-Des)
- Designed-sampled (Des-Mut)
- Train on single mutants (1-vs-rest)
- Train on single and double mutants (2-vs-rest)
- Train on mutants with up to seven changes (7-vs-rest)
- Train on low fifitness, test on high (low-vs-high)
- Sampled:随机划分8:2
Thermostability
Motivation
蛋白质热稳定性在工业上很重要,也是定向进化里很好的一个起点。热稳定性的预测具有挑战性,因为它不一定是一个平滑的功能landscape;在某些蛋白质家族中,一个单一的氨基酸替代可以赋予或破坏热稳定性
Landscape
13个物种48000个蛋白,包括了全局和局部突变体
Splits
- Mixed:对所有序列聚类,使用MMseqs2以20%的序列标识阈值选择聚类代表创建一个split。这个split中的80%的cluster中的所有序列用于训练,剩下的20%测试
- Human:对所有human序列聚类,使用MMseqs2以20%的序列标识阈值选择聚类代表创建一个split,同上
- Human-cell:对所有human细胞系(one cell line for human)序列聚类,使用MMseqs2以20%的序列标识阈值选择聚类代表创建一个split,同上
Baseline algorithms

对于使用蛋白质语言模型的baseline,我们计算每个氨基酸的embedding,用三种方法做pool
- Per amino acid (per AA):pool over the sequence uing a 1D attention layer来做回归预测
- Mean:对整个蛋白质长度每个氨基酸做mean pool
- Mean over subset (mut mean):对mutated region of interst的每个氨基酸做pool
训练的时候,10%随机采样的训练集作为验证集
Results