论文地址:TM-search:An Efficient and Effective Tool for Protein Structure Database Search
TM-search:快速结构比对
Abstract
目前的蛋白质结构搜索算法都需要大量的计算开销,本文设计了TM-search,是基于TM-align的一个新的迭代聚类算法的程序,benchmark显示能比TM-score快27倍同时保持90%的命中率,比其他的现有算法快2-10倍,如foldseek、Dali和PSI-BLAST
Introduction
截至2020年有178,000个结构,~400,000个蛋白质链,这对于TM-align来说计算量太大了,其计算一个蛋白质对需要平均0.5s
现在有两种主流的方法来加速结构数据库检索:
- 将3D结构映射到1D的结构标识符,比如Foldseek等。但这丢失了大量的结构关键信息,也可能导致了其对远端同源检测不敏感
- 通过聚类方法来减少不必要的计算量,成对的结构对齐只在query和representative structures进行计算,像是MMseq2或CD-HIT方法只能减少序列相似度进行聚类,而不能通过结构
TM-search使用基于结构相似度矩阵的层次聚类数据库

Materials and Methods
Preparation of the TM-search Database
创建一个层次结构数据库。
- 每个数据库里的结构拆分成多个domain,如果这个蛋白的domain在SCOPe里面已经定义过了,那么就用这个定义拆分;如果没有,使用Protein Dmomain Parser进行拆分
- 排除了少于30个氨基酸的结构后,获得了~470,000个结构作为初始数据库(PDBall)
- 使用CD-HIT对序列相似度进行70%聚类,每个cluster里面最大的结构作为represent,一共生成了71,115个非冗余数据(PDB70)
接下来针对PDB70进行两两计算TM-score
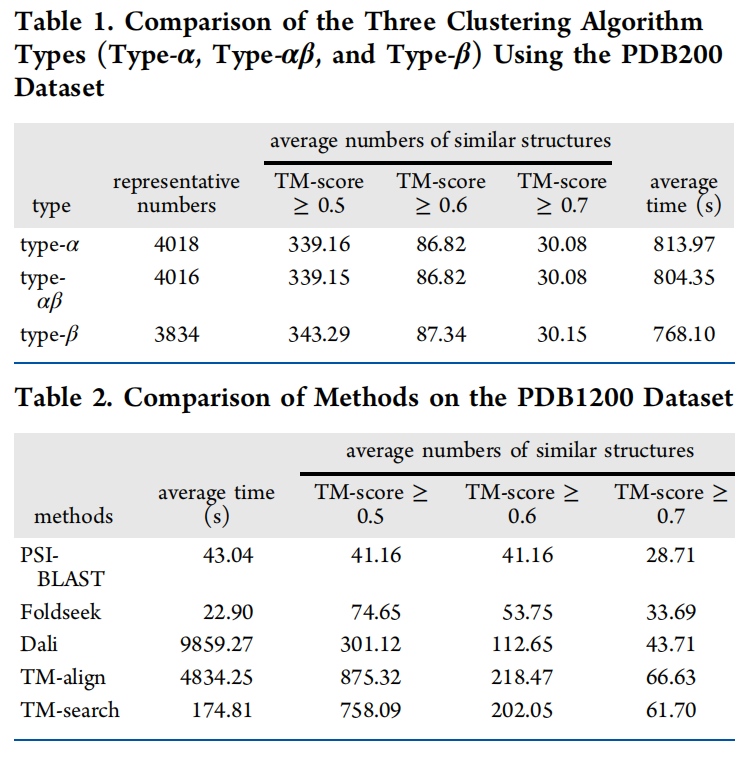
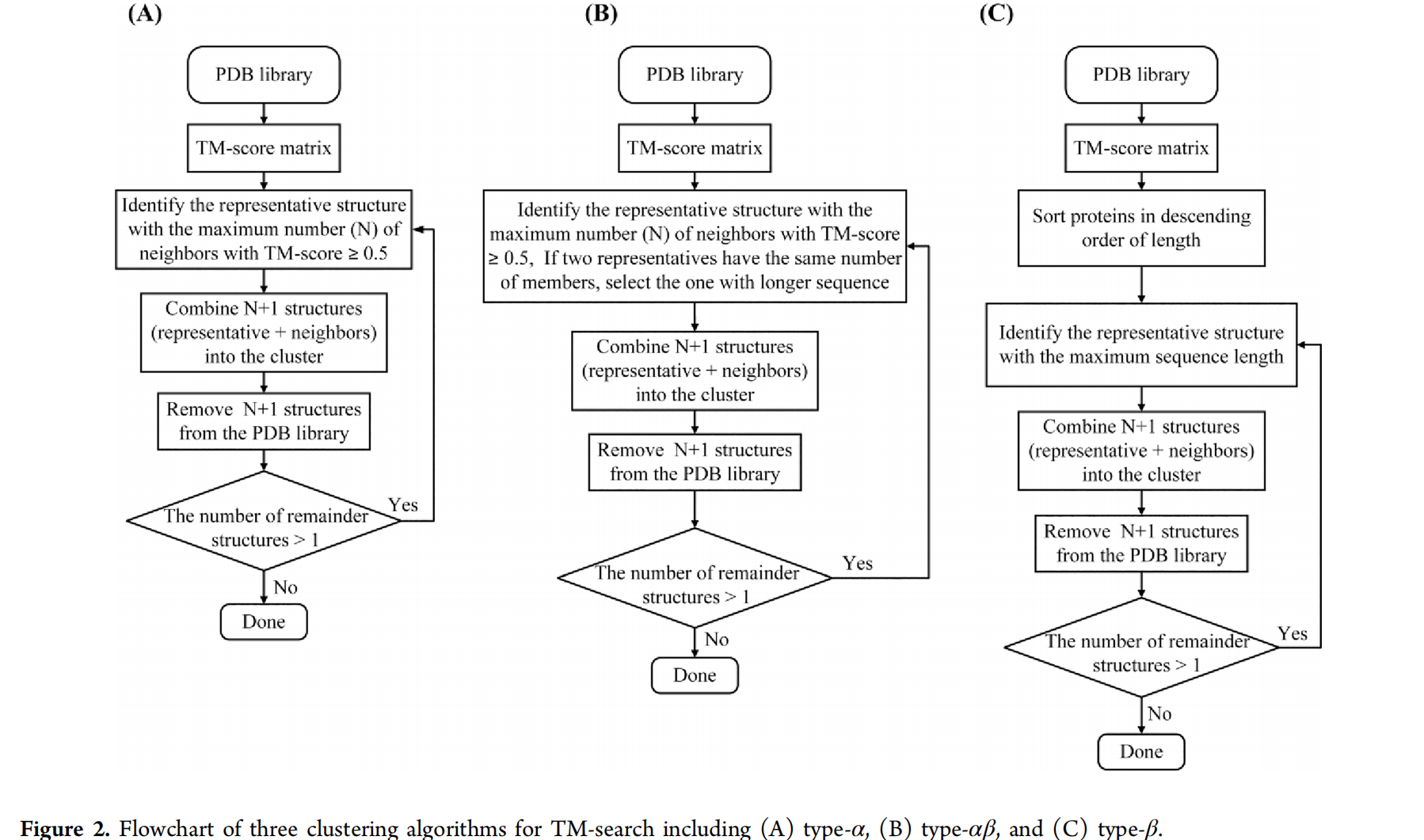
选择聚类代表是影响算法精度和速度的关键,使用了三种不同的策略来选择聚类代表
- type-α:聚类代表是和聚类中TM-score>0.5最多的结构
- type-αβ:跟α差不多,但是如果最后又多个蛋白有相同数量的最大邻居,那么选最长的
- type-β:不属于现存的聚类的最长蛋白作为代表
Evaluation Metric
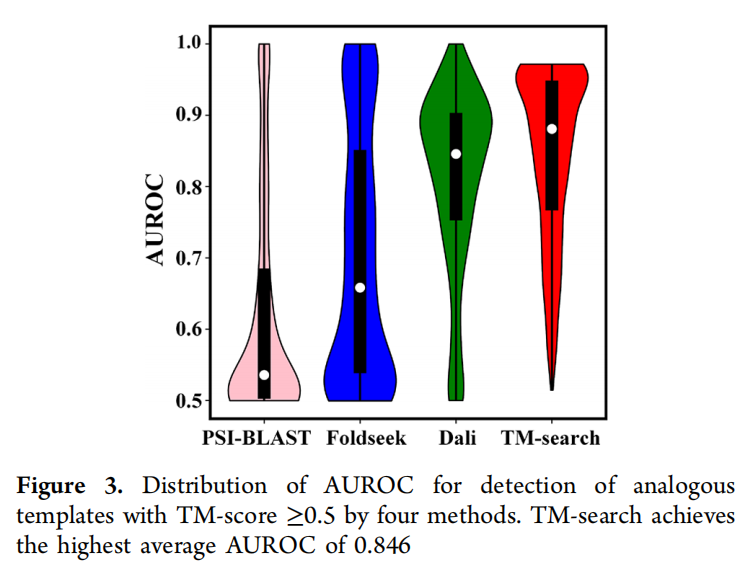
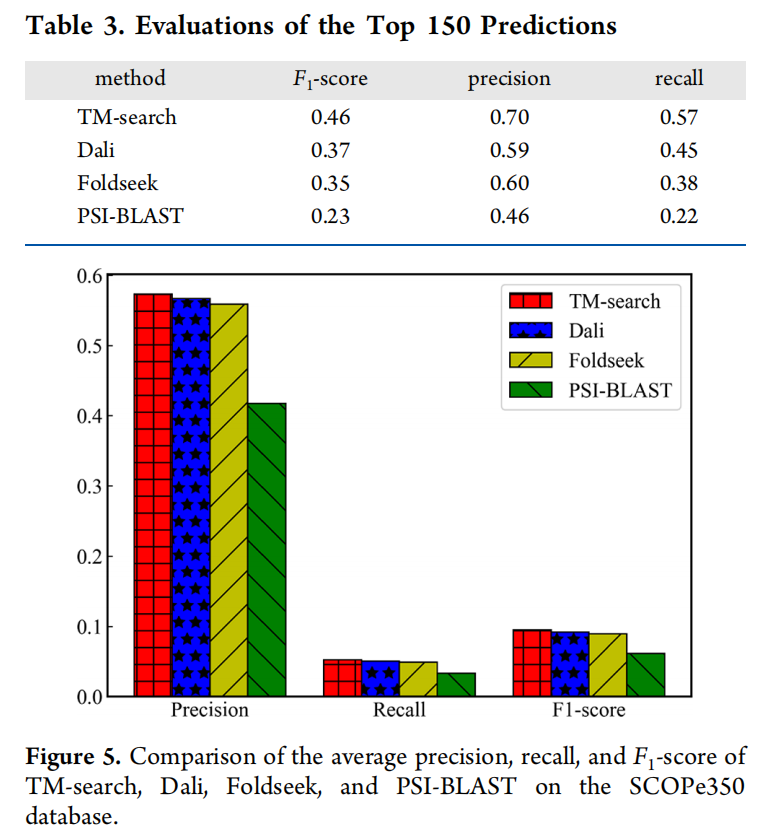
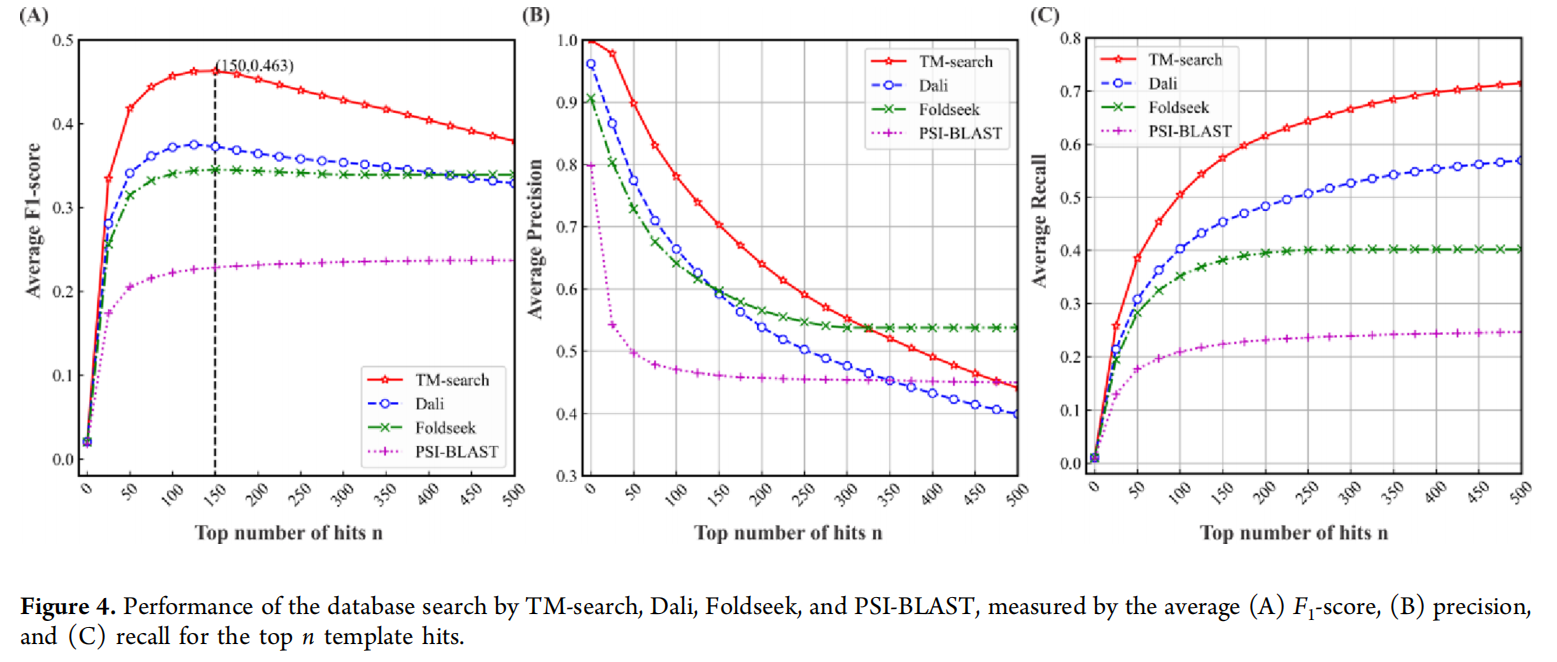
AUROC、F-score、recall、precision
Algorithm for Database Search
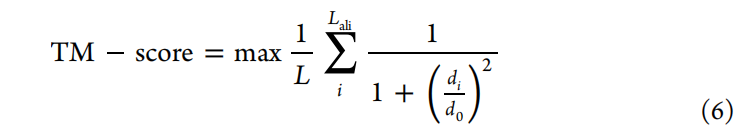

TM-score
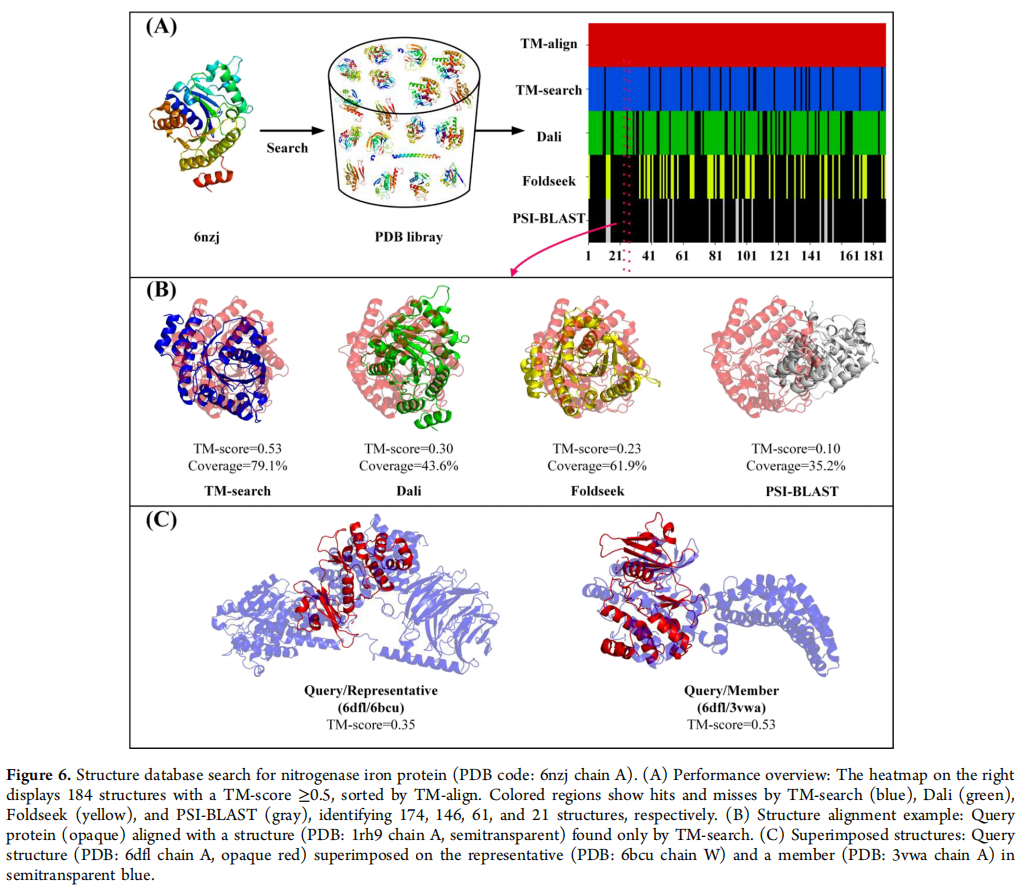
Results and Discussion