论文地址:Fast and accurate protein structure search with Foldseek
Foldseek:结构序列化+快速结构比对
Abstract
Foldseek通过将蛋白质内的三级氨基酸相互作用描述为结构字母表上的序列,将查询蛋白质的结构与数据库对齐。将计算时间减少了4到5个数量级,相较于Dali,TM-align和CE分别有86%,88%和133%的灵敏度
Introduction
最广泛使用的蛋白质注释和分析方法是基于序列相似性搜索,目的是找到同源序列,从中可以推断出查询序列的属性。尽管基于序列的同源性推理取得了成功,但许多蛋白质无法进行注释,因为仅从序列中检测遥远的进化关系仍然具有挑战性。尽管几十年的努力来提高结构对准器的速度和灵敏度,目前的工具在处理当今结构数据库规模方面太慢了
在一亿蛋白质结构数据库上查询一个蛋白,TM-align单核需要一个月,如果做all-versus-all全面比较需要1000核集群一万年;用序列搜索的方法,MMseqs2只需要一个星期
结构对齐工具慢有两个原因:
- 序列对齐有prefilter算法来减少计算的数量级,结构对齐没有
- 结构相似度是非局部的,改变一个部分的对齐会对其他部分有相似性的影响,目前的方法基本都是采用迭代或随机优化来解决
为了加速可以把蛋白质的氨基酸骨架描述为序列,然后用序列对比方法来比较结构,所以结构字母表的方法可以提速
对于Foldseek,我们开发了一种结构字母表,它不描述蛋白质的骨架,而是描述蛋白质的三级结构相互作用。3D相互作用(3Di)字母表的20个状态描述了每个残基i与其空间上最近的残基j之间的几何构型。与传统的骨架结构字母表相比,3Di具有以下三个主要优势:
- 连续字母之间的依赖性较弱:3Di字母之间的依赖性较弱,这意味着每个残基的状态更多地受其局部环境的影响,而不仅仅是前一个残基的状态。这种较弱的依赖性有助于提高信息密度,并减少误报的概率。
- 状态频率更均匀分布:3Di字母表中的状态频率更均匀分布。相比之下,传统的骨架结构字母表中,一些状态的频率可能会比其他状态更高,导致信息的不均衡。而3Di字母表的均匀分布有助于提高信息密度并减少误报的概率。
- 信息密度分布:最高的信息密度编码在保守的蛋白质核心区域,而最低的信息密度编码在非保守的卷曲/环区域。与此相反,对于骨架结构字母表,其信息密度分布与3Di相反。这意味着通过3Di字母表,我们可以更好地捕捉蛋白质保守区域的结构信息,而在非保守的卷曲/环区域提供较少的信息
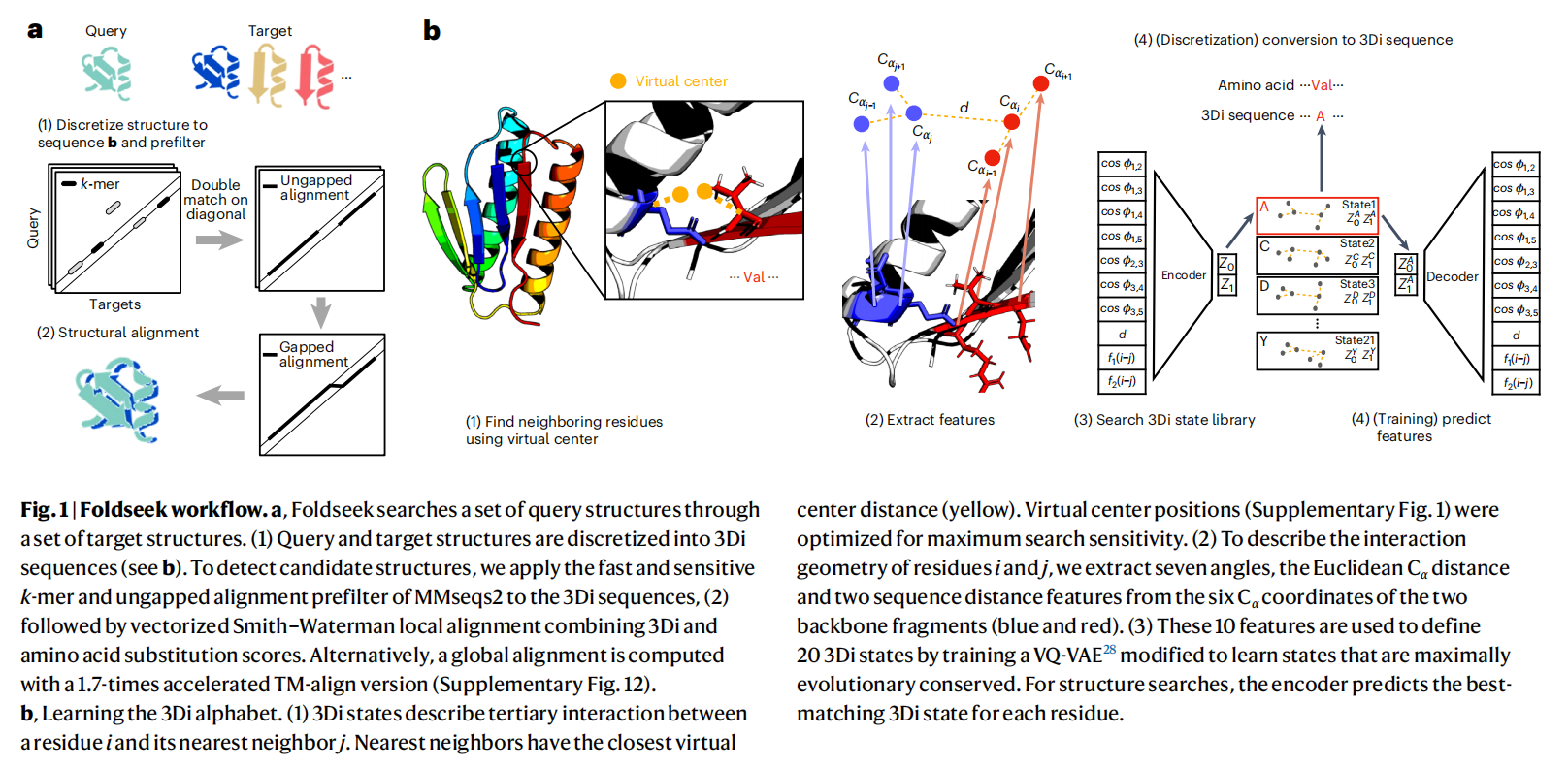
- 将查询结构离散化为基于3Di字母表的序列:Foldseek将查询结构转化为基于3Di字母表的序列。这个字母表描述了蛋白质结构中残基之间的三维相互作用。3Di序列通过表示每个残基与其空间上最近的残基之间的几何构型来描述。
- 利用预训练的3Di替代矩阵进行搜索:Foldseek使用预训练的3Di替代矩阵来搜索目标结构库中的3Di序列。搜索过程利用了MMseqs2软件的双对角线k-mer预过滤和无间隙对齐预过滤模块。得分较高的匹配结果通过局部对齐(默认使用3Di)或全局对齐(Foldseek-TM)进行进一步处理。局部对齐阶段结合了3Di和氨基酸替代得分。3Di字母表的构建过程可以参考论文中的图1b和补充图1-3。
- 降低误报率并计算可靠的E值:为了减少高分误报和提供可靠的E值,Foldseek采取了以下策略:通过从原始得分中减去反向查询对齐得分来消除假阳性;在局部40个残基的序列窗口内应用组成偏差校正。E值使用基于极值分布的得分分布进行计算,其中极值分布的参数是基于3Di序列组成和查询长度预测的神经网络计算得到的。匹配结果的排名由对齐比特分数乘以对齐TM分数和局部距离差异测试(LDDT)的几何平均值确定。
- 提供匹配的同源概率和排序:Foldseek根据SCOPe数据库上真实和假匹配的对比来估计每个匹配的同源概率。匹配结果按照对齐比特分数乘以对齐TM分数和局部距离差异测试(LDDT)的几何平均值进行排序。
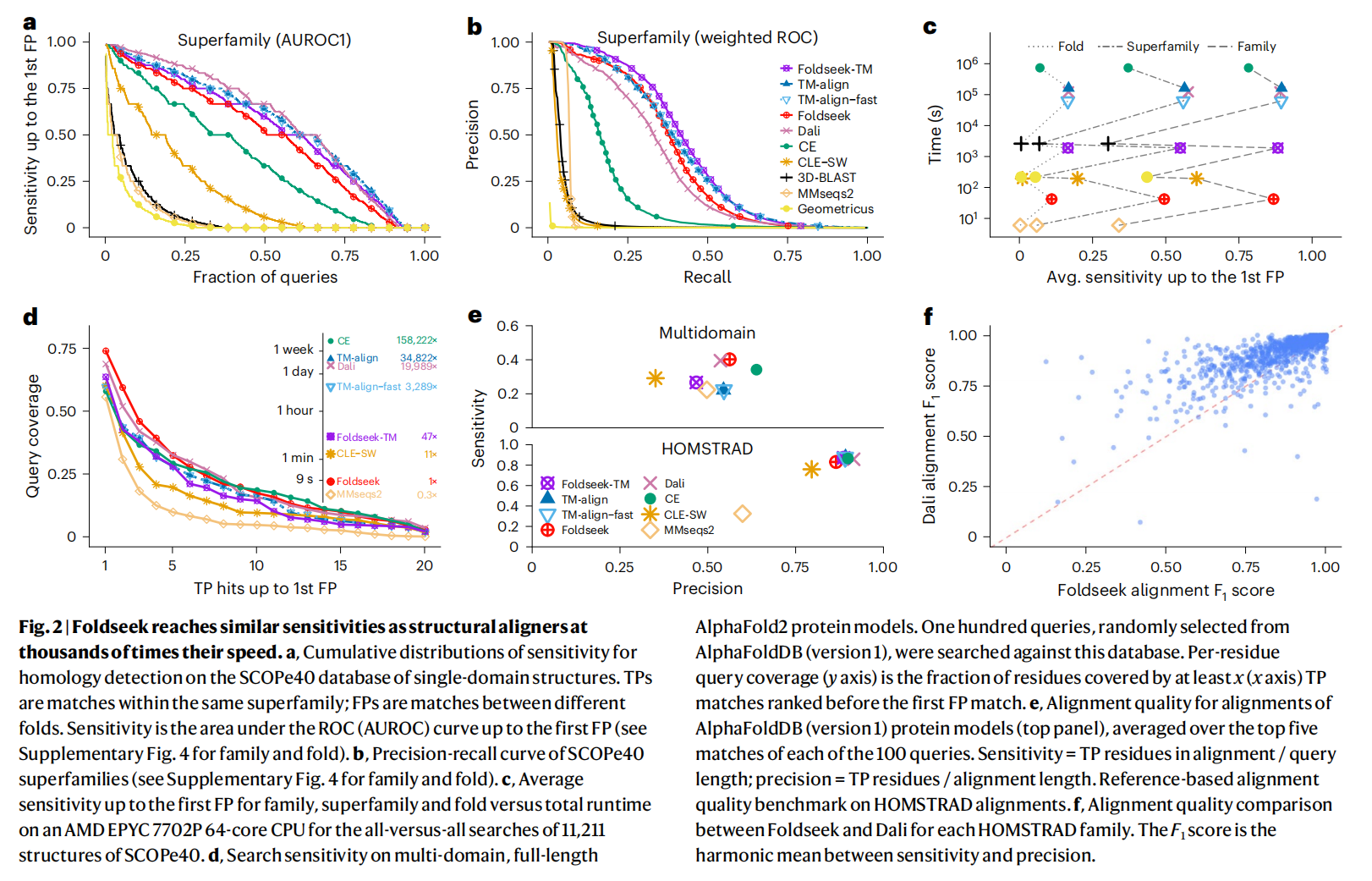
Foldseek在蛋白质结构比对中表现出较高的灵敏度和精确度,并且具有显著的计算速度优势。与其他常用的结构比对工具相比,Foldseek在家族和超家族级别的关系检测中具有更好的性能,并且在速度上明显快于传统的工具
Methods
Overview
Foldseek的核心模块建立在MMseqs2框架上,利用了以下几个关键技术来实现高性能:
- 3Di序列比对:Foldseek通过使用3Di字母表对结构进行编码,并将结构比对转化为3Di序列比对。这种基于3Di的序列比对方法能够更准确地捕捉结构之间的相似性。
- 高效的预过滤器:Foldseek的预过滤器能够高效地筛选出具有高相似性的候选比对。预过滤器通过寻找在动态规划矩阵的同一对角线上的两个相似的、间隔的3Di k-mer匹配来实现。这种非精确匹配的策略在提高灵敏度的同时大大减少了需要进行完整比对的序列数量。
- 多线程和SIMD优化:Foldseek利用多线程和SIMD向量单元来提高计算性能。多线程技术允许同时处理多个任务,加快计算速度。而SIMD向量单元可以一次执行多个相似操作,进一步提高效率。
Descriptors for 3Di structural alphabet
本文开发了一种不描述骨架,而是描述三级相互作用的结构字母表。三维相互作用(3Di) 字母表的20个状态描述了每个残基i与其空间上最近的残基j之间的几何构型。与传统的蛋白质骨架结构字母表相比,3Di具有三个关键优势
- 连续字母之间的依赖性较弱
- 状态频率更均匀分布,这两者都增强了信息密度并减少了假阳性(FPs)
- 最高的信息密度编码,在保守的蛋白质核心中,而最低的信息密度编码在非保守的螺旋/环区域中,而对于骨架结构字母表则正好相反。
Foldseek
Foldseek主要有两个步骤:
-
将待比对的蛋白结构离散化为3Di字母表上的序列,然后使用预先训练好的3Di替代矩阵,利用MMseqs2中基于双对角k-mer的预过滤器和无缝比对预过滤器模块,搜索目标结构的3Di序列
-
高得分匹配使用3Di (默认)在本地对齐,或者使用TM-align在全局对齐(Foldseek-TM) 。本地对齐阶段将3Di和氨基酸替代分数进行组合

