论文地址:Protein remote homology detection and structural alignment using deep learning
TM-vec/DeepBLAST:TM-align训练的检索和比对模型
Abstract
在低相似度的情况下探索蛋白质序列-结构-功能关系现在需要改进方法,因此提出了TM-vec和DeepBLAST。前者可以在序列层面实现结构-结构相似度的计算,因为是在TM-score的数据库上进行训练;后者可以只使用序列实现在结构上的对齐。它优于传统的序列对齐方法,其性能类似于基于结构的序列对齐方法,文章展示了TM-Vec和DeepBLAST在各种数据集上的优点,包括与最先进的序列比对和结构预测方法相比,能够更好地识别远程同源蛋白
Introduction
根据序列相似度来检测蛋白质的同源性是一个标准方法,过去50年里被使用到了蛋白质注释预测、预测蛋白质结构、蛋白质相互作用、设计蛋白质和进化关系建模上
大多标准的序列同源方法依赖于高序列相似度,相较于序列同源,结构同源在时间尺度上保留得更好。由于进化关系的遥远,超过一半的蛋白质在标准序列数据库中没有序列同源性。近期宏基因组的分析表示在使用结构同源检测后注释率可以达到70%以上。对密切相关的蛋白质使用基于序列比对的方法,对远缘相关蛋白使用基于结构比对的方法可能是一种理想的杂交方法,提供更好的灵敏度
尽管像TM-align,Dali,FAST和Mammoth等方法可以在低序列相似度下提供结构度量,但是有两个主要的局限:
- 大多蛋白质没有蛋白质结构
- AlphaFold2对于短蛋白质预测能力有限

由于结构数据库的快速增长,现有的结构比对方法无法扩大到这么大规模上
开发了可以对蛋白质结构进行微调的神经网络
- 使用孪生神经网络预测蛋白质对之间的tm分数
- 使用可微的 Needleman–Wunsch algorithm预测蛋白质之间的结构对齐
Results
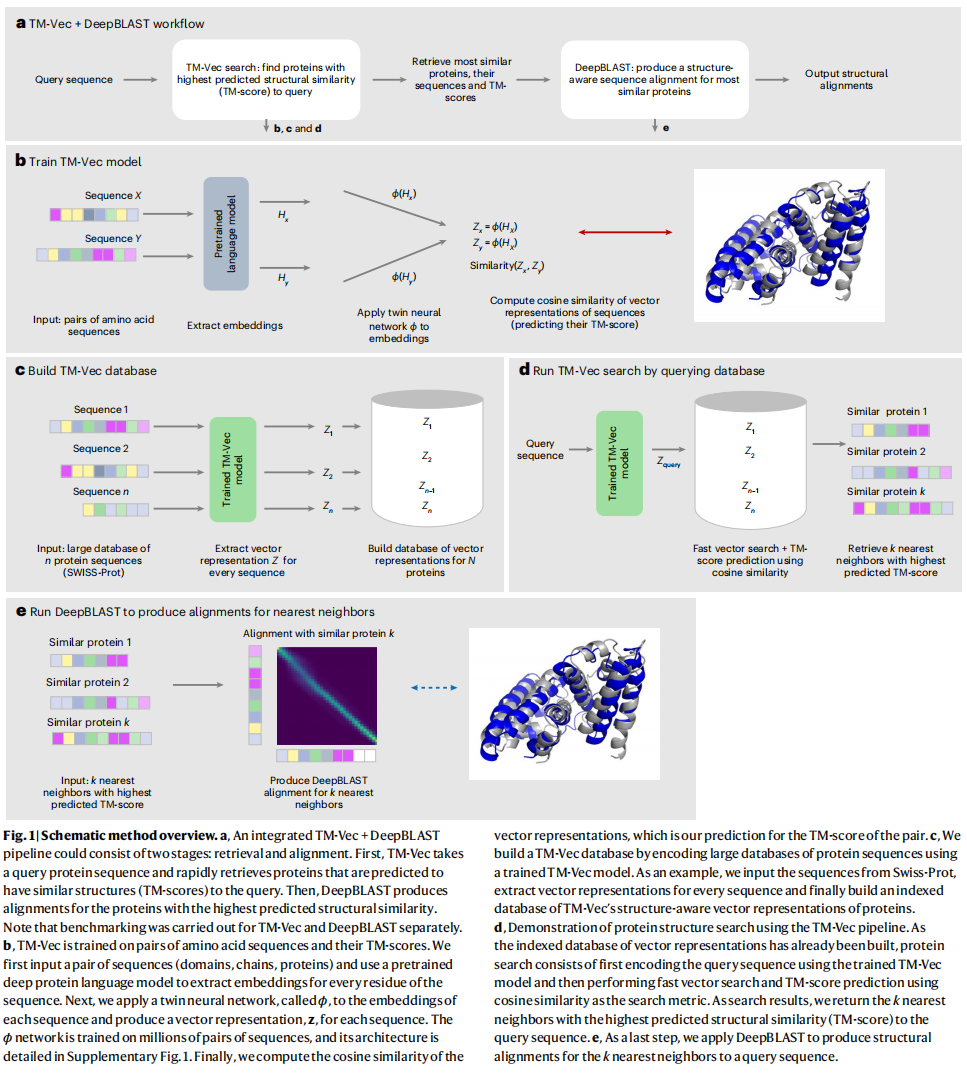
在Malidup和Malisam结构数据库上展示了DeepBLAST的结构aligment能力,在CATH、SIWSS-MODEL、Malidup和Malisam上展示了TM-vec的能力
Scalable structural alignment search using neural networks
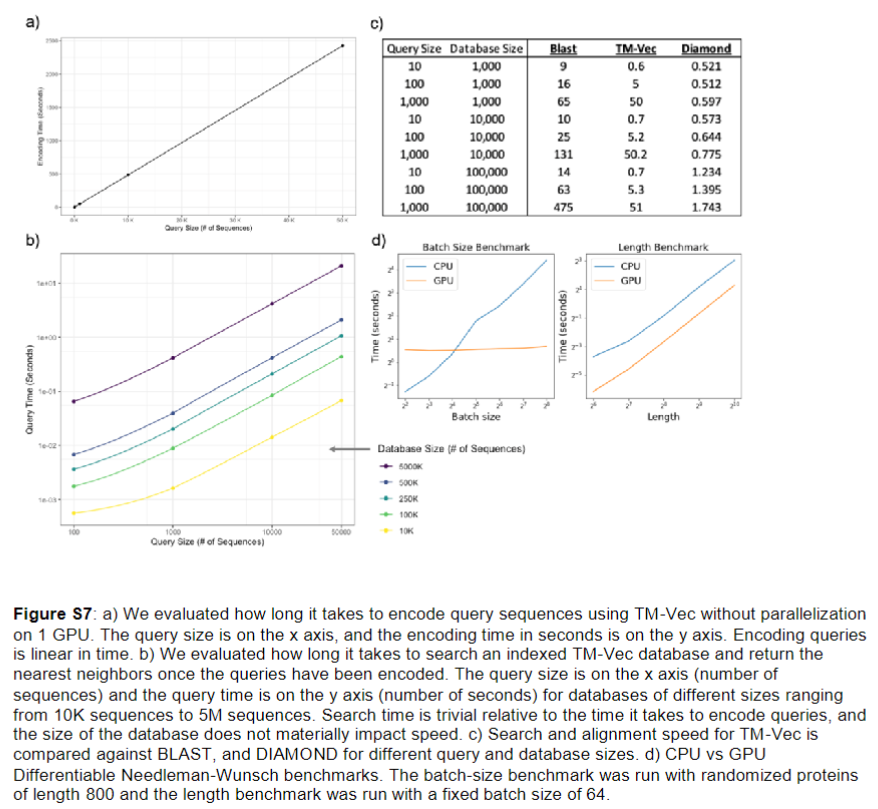
直接使用DeepBLAST是随着数据库线性增长的,这是不可取的,而使用TM-vec编码成向量之后对于n个蛋白的查询复杂度就变为了O(log^2n)
SWISS-MODEL进行了大约150M的蛋白质对训练,还有在CATH 40%序列相似度的数据上进行训练
为了进一步验证TM-vec模型,还在Microbiome Immunity Project (MIP)上进行了验证,它包含了20万个来自先前未知蛋白质的从头蛋白质结构,包括148个假定的折叠,和TM-score的相关系数达到了0.785
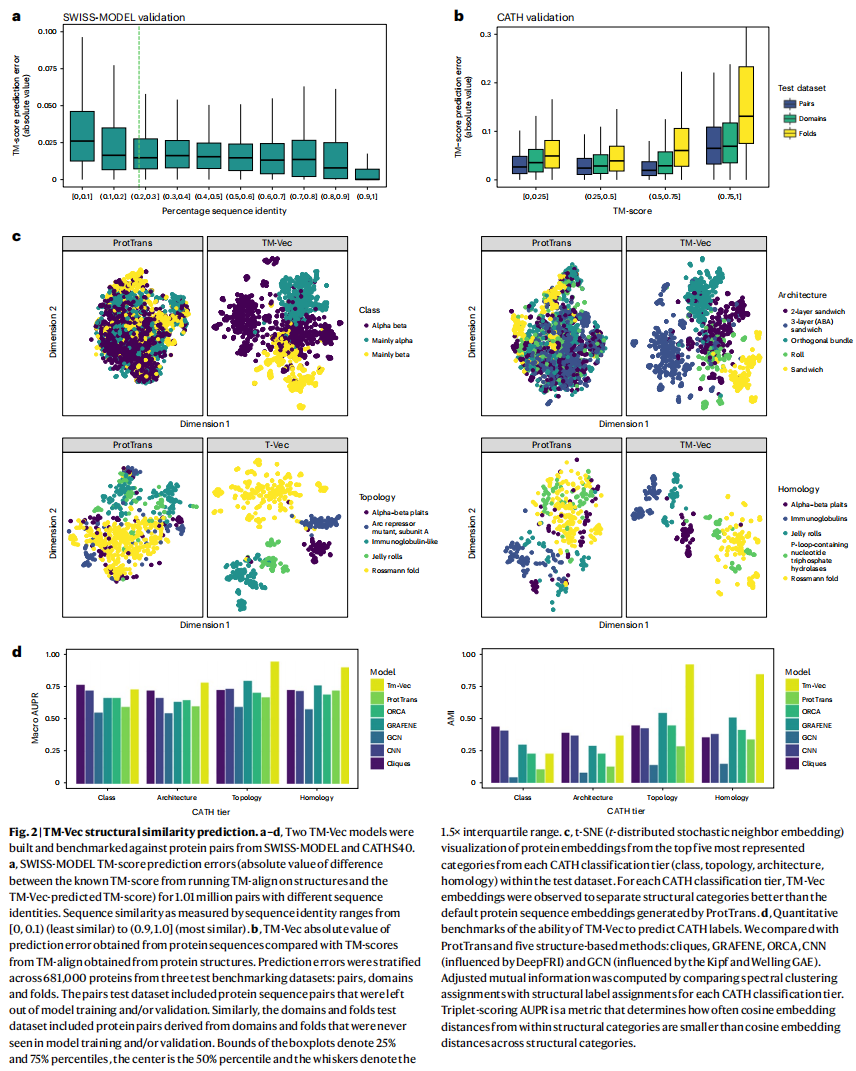
Capturing structural information in the latent space
图2c是可视化结构
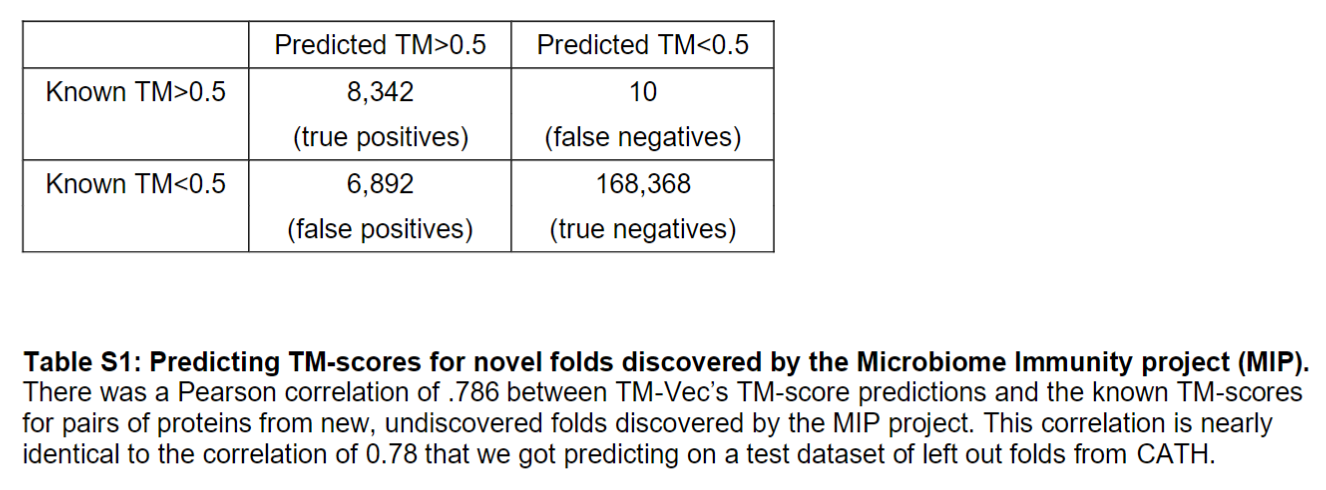
针对Microbiome Immunity project (MIP) 新颖 Fold 数据集,TM-Vec 在判定2个蛋白是否具有相同 Fold 方面,假阳率(0.1%)和假阴率(3.9%)都不高
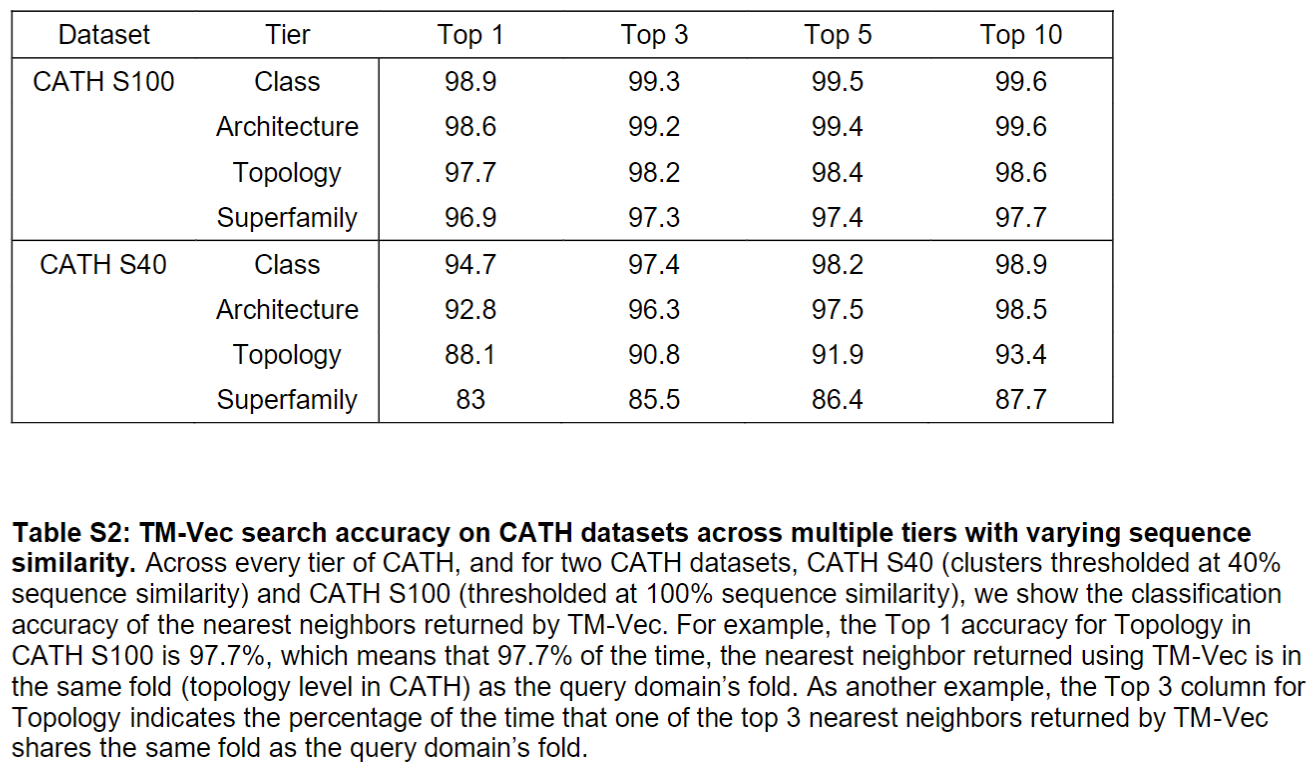
给定query蛋白,可以在CATH上达到很高的准确率
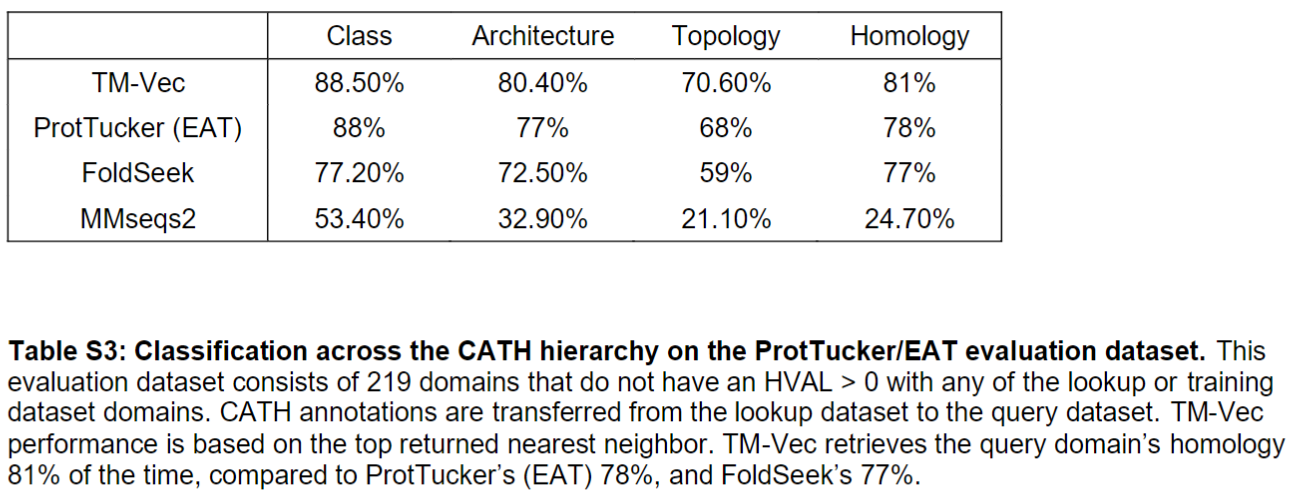
为了和其他Baseline有头对头的比较,ProtTucker 基准测试集的219个结构域为测试集,在81%的情况下,TM-Vec 正确地返回了 query 序列的 homology,而 Foldseek 则是77%

因为219个domain太少了,在更大的 CATH S20 上测试,在 CATH 上训练的 TM-Vec 同样在搜索 CATH homology 方面有最好的表现,优于基于结构搜索的 Foldseek 和基于序列搜索的 HHBlits、 MMSeqs2 等
Extracting structural alignments from sequence
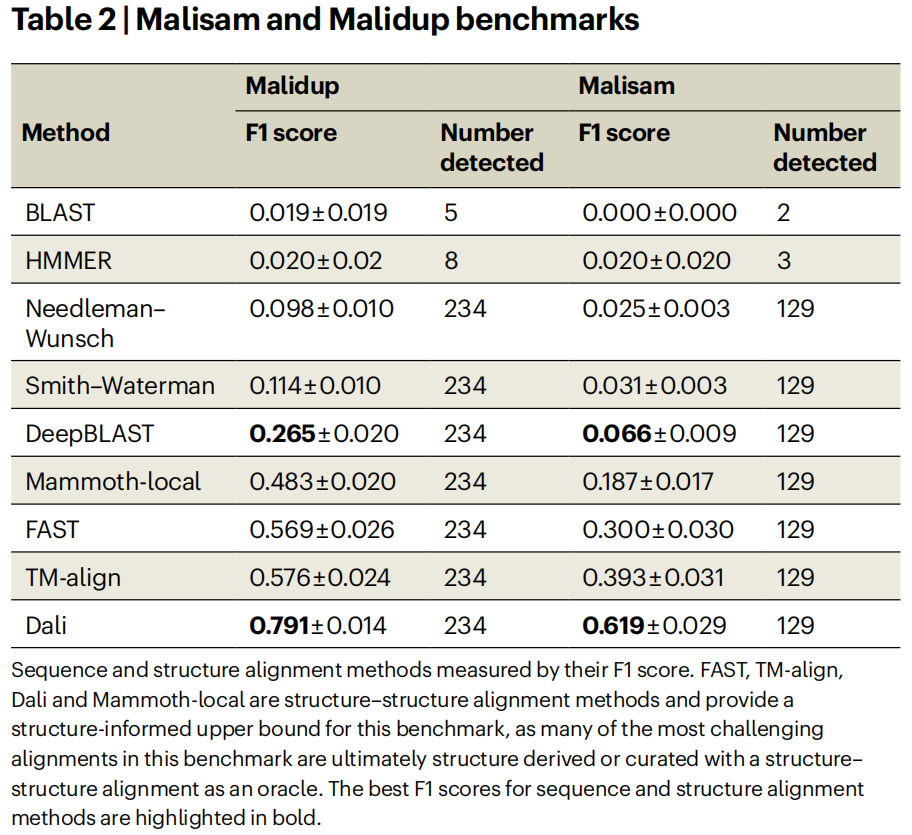
DeepBLAST只使用序列,没有提供任何的原子坐标,使用了两个benchmark named Malisam and Malidup
- Malidup(Proteins, 2003)的全称是 Manual ALIgnments of DUPlicated domains,含241组手动对齐的复制结构域,是首个不基于序列但结构相似的 homolog 结构库。这里的 duplicated domains 指一些蛋白含有多个重复的结构域,这些结构域尽管同源,但往往执行不同的功能,并在序列上有较大分化,因此 Malidup 内的数据是低序列相似度但高结构相似度的同源结构域
- Malisam(NAR, 2007)的全称是 Manual ALIgnments for Structrually Analogous Motifs,含130对手动对齐的结构类似模体,是首个 structural analog 数据库
- 手动对齐过的蛋白结构,可以认为实现了最佳对齐,缺点是数据库太老、太小。但是,其优点也是显然的:对其中数据使用序列对齐方法,如 BLAST 和 HMMER,难以将之对齐(大多数无法通过阈值过滤检测)
Remote homology detection and alignment
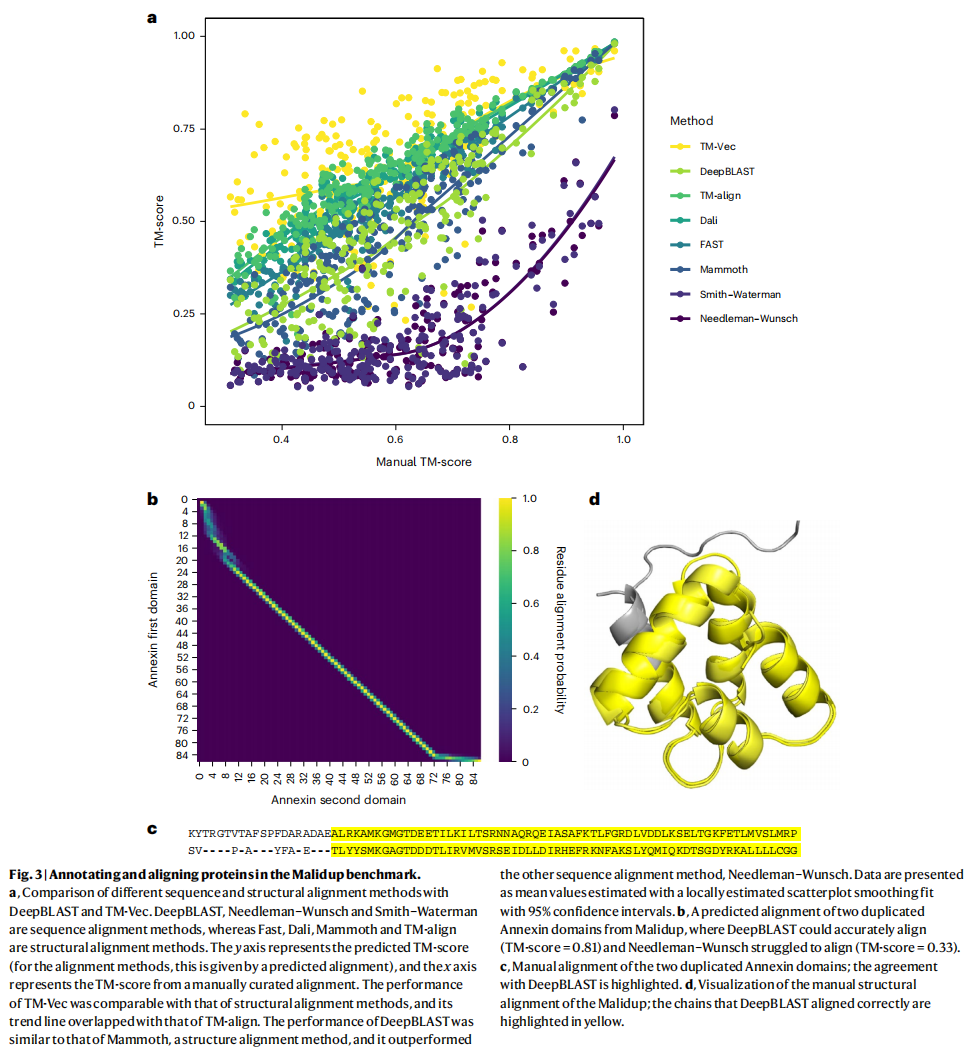
Malidup数据集,具有明显的结构相似区域,人工建立的结构-结构aligment,让序列aligment方法无法检测的低序列相似度
图(b)是每个位点的对齐概率,(c)是这两个小蛋白的对齐的序列,可见相似度是比较低的;(d)是膜联蛋白(annexin)的两个结构域,DeepBLAST 将之对齐后计算 TM-score 为 0.81,而使用 Needleman–Wunsch 先对齐序列再按对齐的序列去计算对齐的(部分)结构,得 TM-score 为 0.33
Full repository-level scaling and runtime
Case study: bacteriocins
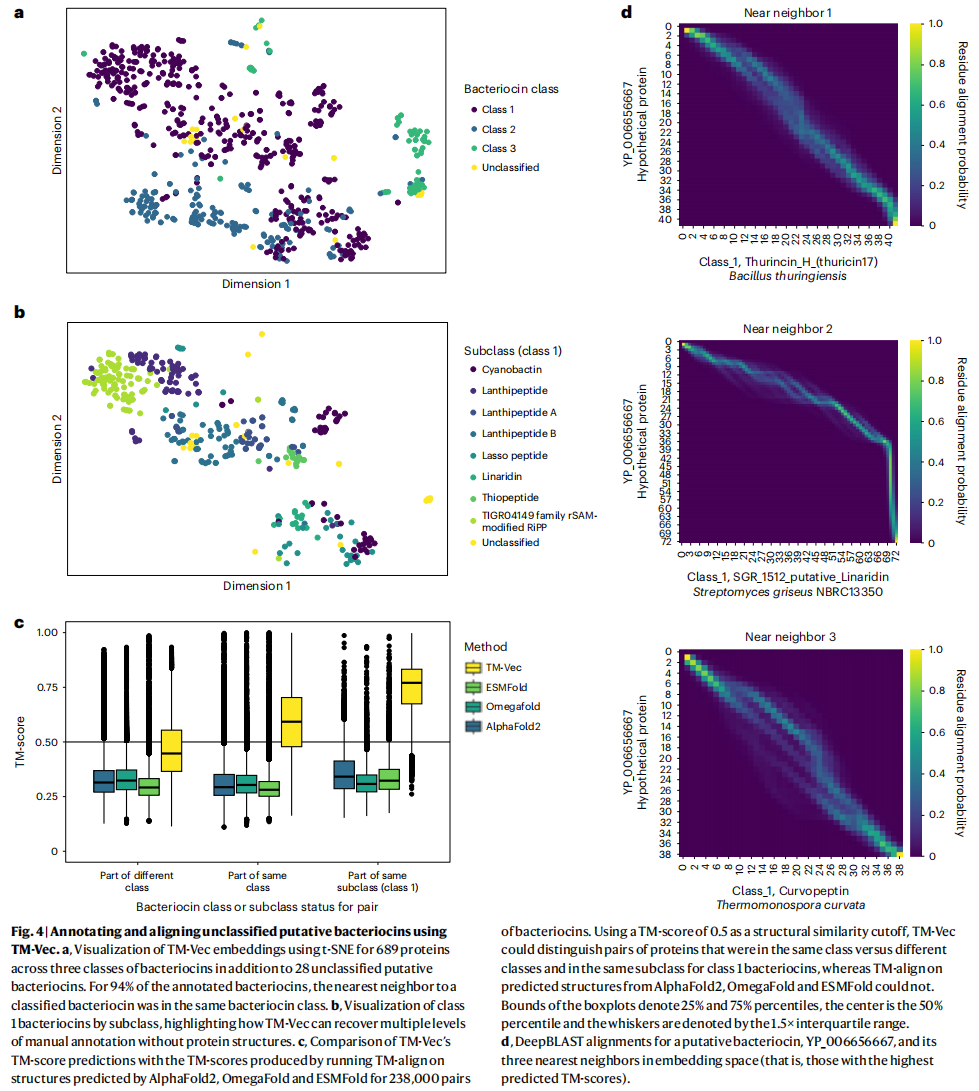
细菌素 bacteriocin 是结构、序列均非常多样的多肽(长度一般小于50),难以基于现有的序列同源检测工具挖掘。另一方面,细菌素广泛存在于各类细菌中,考虑到庞大的细菌种类,现在仅识别出的约1000种细菌素太少了,使用BAGEL database分析了细菌素家族的多样性。也就是说,在蛋白组、宏基因组等数据中侦测识别细菌素很困难。
TM-Vec 对未标注细菌素的区分效果,比较对象是 AlphaFold2, OmegaFold, ESMFold 的预测结构。具体而言,作者用三种 Fold 分别预测这些细菌素的结构,分别对同属一个类型和分属不同类型的细菌素计算预测结构之间的 TM-score。图4:三种 Fold 预测出的结构,互相比较,无论是否属于同一类,其 TM-score 中位数均在0.3 左右,无法区分;然而,TM-Vec 却能进行区分
Discussion
- TM-vec无法检测出点突变的结构不同
- 因为是预测tm score,这是衡量结构全局相似度的,无法对局部相似度进行判断
Methods
TM-Vec search
TM-Vec embedding model
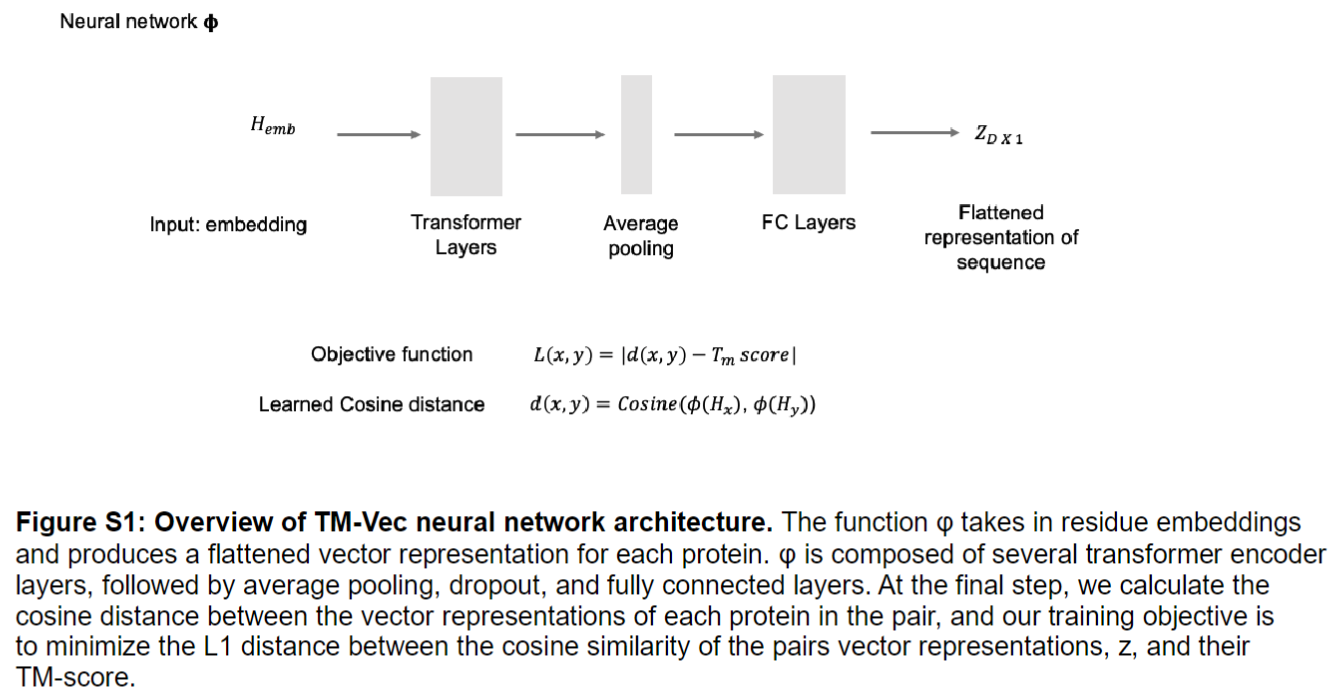
使用的PLM是ProtTrans (ProtT5-XL-UniRef50)
TM-Vec database creation
使用的FAISS构建向量库
DeepBLAST alignment module
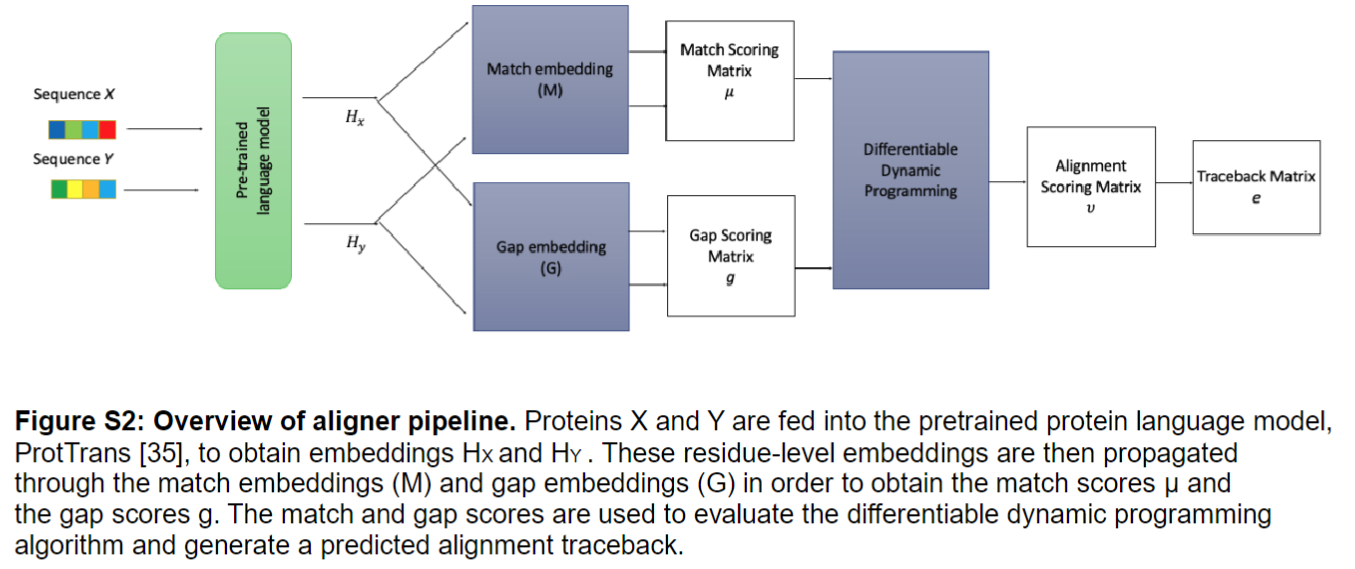
使用 differentiable Needleman–Wunsch algorithm
把经过PLM的residue-level的embedding通过一个match embeddings (M)和一个gap embeddings (G)得到match score μ和gap socre g,然后经过一个动态规划算法得到一个alignment traceback,这些alignments可以用真实的alignments进行微调
Protein language modeling for alignment
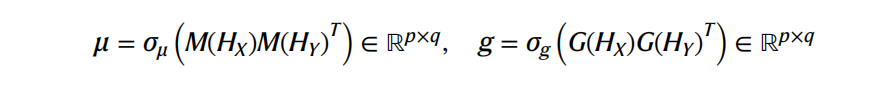
p和q表示蛋白质X和Y的长度,d为语言模式的嵌入维数
Differentiable dynamic programming
这里的微分动态规划框架没有任何可学习参数,只是为了让M和G能够反向传播
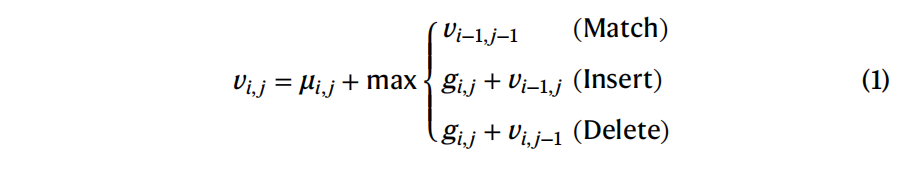
alignment score $v_{i,j}$ 是第一条序列的第i个和第二条序列的第j个,$\mu_{i,j}$ 表示氨基酸 $X_i$ 和 $X_j$ 对齐了的log-odds socre,$g_{ij}$ 表示插入 或删除的log-odds score,$v_{n,m}$ 是两条序列的最佳对齐分数,通过argmax操作,通过对齐矩阵跟踪得分最高的路径,可以得到最优对齐
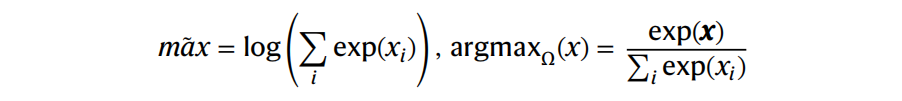
因为max和argmax操作都不是可微的,所以使用了一个平滑的操作
Alignment loss function
使用预测值和ground truth alignments的欧氏距离作为loss
Datasets
Protein-chain-pairs dataset
从SWISS-MODEL里面采样paris of chains,里面有超过500,000 chains
对于chain-pairs数据集,最多300氨基酸长度的训练/验证集有141M对,held-out测试集是1M对,超过1000氨基酸的有320M对
Domain-pairs dataset
丢掉了超过300个氨基酸的domain,得到450M对,再对来自不同fold的CATH domains进行了欠采样,最终得到23M对
Structure alignment dataset
用TM-align从PDB获取了1.5M的alignments用于训练DeepBLAST,包含40,000蛋白结构
ProtTucker benchmark dataset
训练和查找数据集分别由66,000个和69,000个CATH域组成。测试数据集不包括任何具有HSSP值>0的域和任何查找域,并由219个域组成
DIAMOND benchmark dataset
包含了单domain和多domain的蛋白
TM-Vec training
- 在CATHS40和SWISS-MODEL最长300的数据集上训练的模型17.3M参数
- 在CATHS100和SWISS-MODEL最长1000的数据集上训练的模型34.1M参数
学习率1e-4,batch size 32
DeepBLAST training
8个维数为1,024的卷积层来参数化match emebdding M和gap embedding G,最终的模型有1.2B参数,学习率5e-4,batch size 360,在24张A100上训练20 epochs for 6 days

