论文实现:https://git.scicore.unibas.ch/schwede/EBA and https://git.scicore.unibas.ch/schwede/eba_benchmark
EBA:PLM embedding相似度相似度矩阵作为NW和SW的输入
Abstract
Motivation
蛋白质语言模型能够生成per-residue的高维表征,也就是蛋白质序列的语义信息,可以用于下游任务,也可以用于找到蛋白质之间的同源关系
Results
提出了 embedding-based protein sequence alignments (EBA),甚至可以在twilight zone捕获到结构相似性
Introduction
标准的比对方法当进入twilight zone的时候,这种成对信号就会变得模糊,但PLMs建立序列之间的同源关系超越了简单的序列比较,可以揭示更多信息
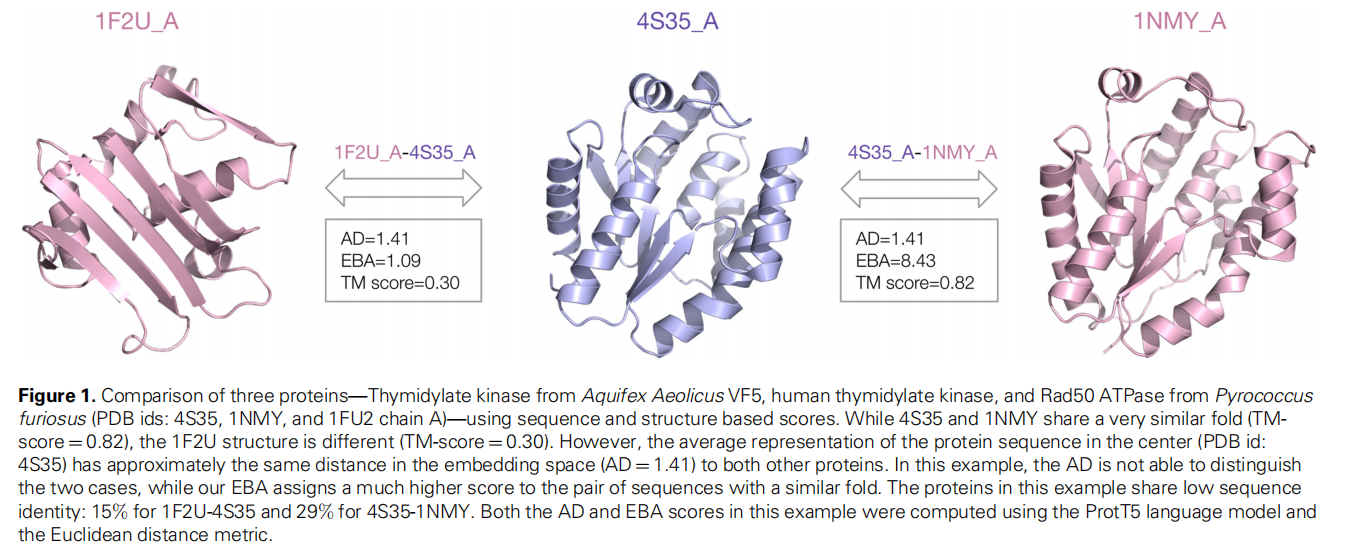
把表征做平均会丢失很多顺序信息,比如图1里面结构、序列完全不一样的蛋白他们的平均表征计算得到的欧几里得距离是一样的
这种问题可以通过增加计算复杂度、显式建模对齐来解决,由于残基水平的嵌入并不总是具有可比性,还包括了一个步骤,使用比较蛋白质的残基嵌入距离的分布来增强相似矩阵的信号
Materials and Methods
benchmark包含了三种预训练模型,ProstT5、ProtT5和ESM-1B
Average distance
把向量平均(AD)算相似度
Embedding-based alignment
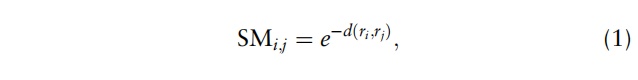
d是欧几里得距离,$r_i$ 和 $r_j$ 分别是两个蛋白质的对应点位的embedding
Signal enhancement
对于给定的一对氨基酸 (i, j) 和相似度分数 $SM_{i,j}$ ,计算同一行或同一列的Z-score
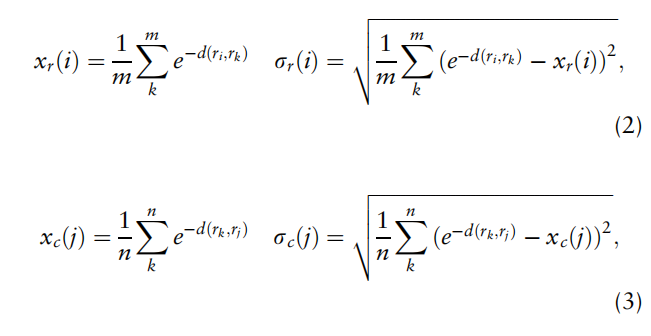
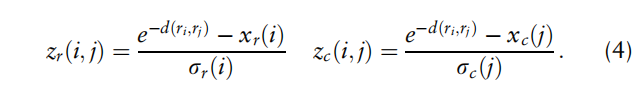
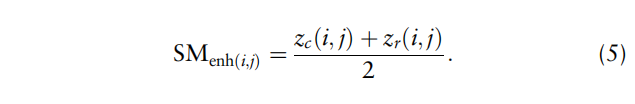
Global and local dynamic alignment
使用 $SM_{enh}$ 作为打分矩阵,Needleman-Wunsch (NW) 和 Smith-Waterman (SW)方法进行比对,gap penality对于NW是0,对于SW是2。对于NW global alignment,EBA similarity socre用下面的方法正则化:
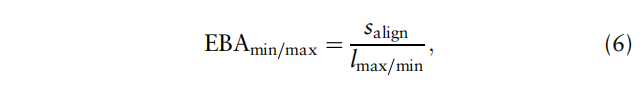
Benchmark
Structural similarity analysis
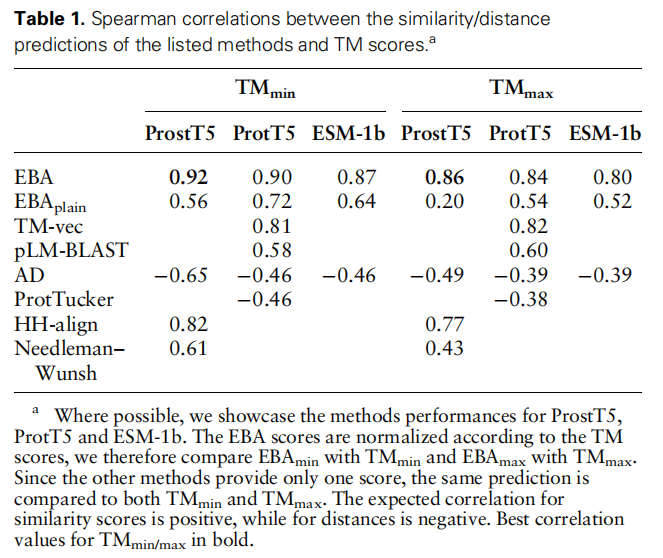
使用PISCES,序列相似度小于30%,最长链小于75,用和tm score的spearman相关系数表示
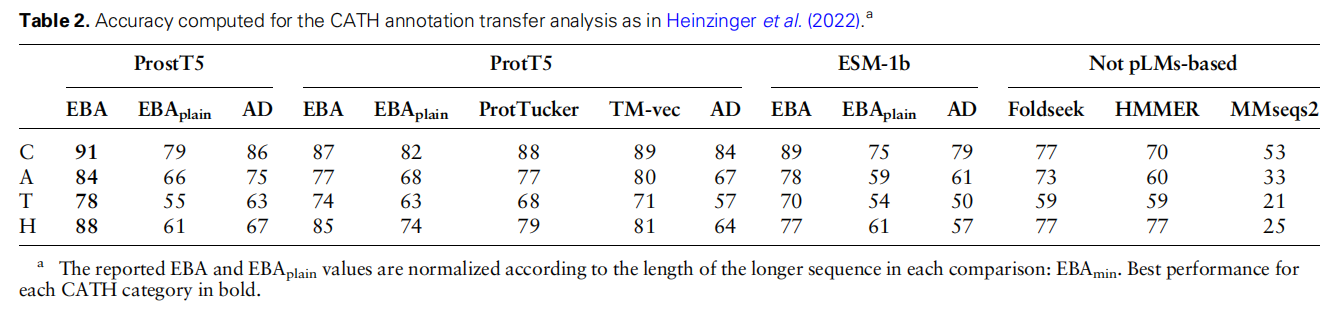
CATH annotation transfer analysis
66K CATH 结构域,219个测试集,用准确率表示
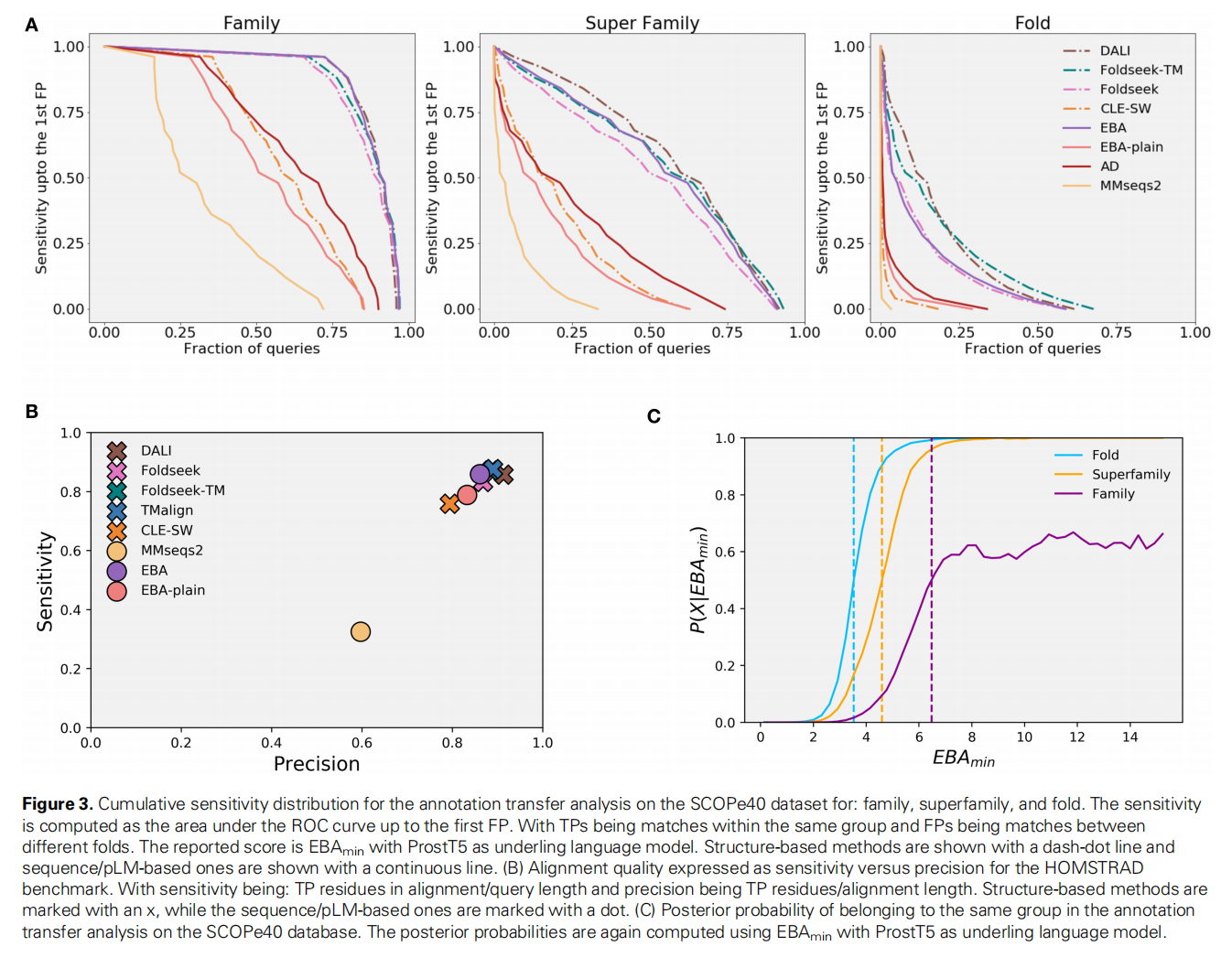
SCOP annotation transfer analysis
SCOPe 2.01中的蛋白结构域聚类在40%的序列同源性,得到12,211个非冗余结构域,数据来源 https://github.com/steineggerlab/foldseek-analysis
HOMSTRAD alignment quality
HOMSTRAD 包含了1032个蛋白家族中的同源蛋白的专业策划的结构比对,数据来源 https://github.com/steineggerlab/foldseek-analysis
Results
EBA captures structural similarity in the twilight zone
Length normalization
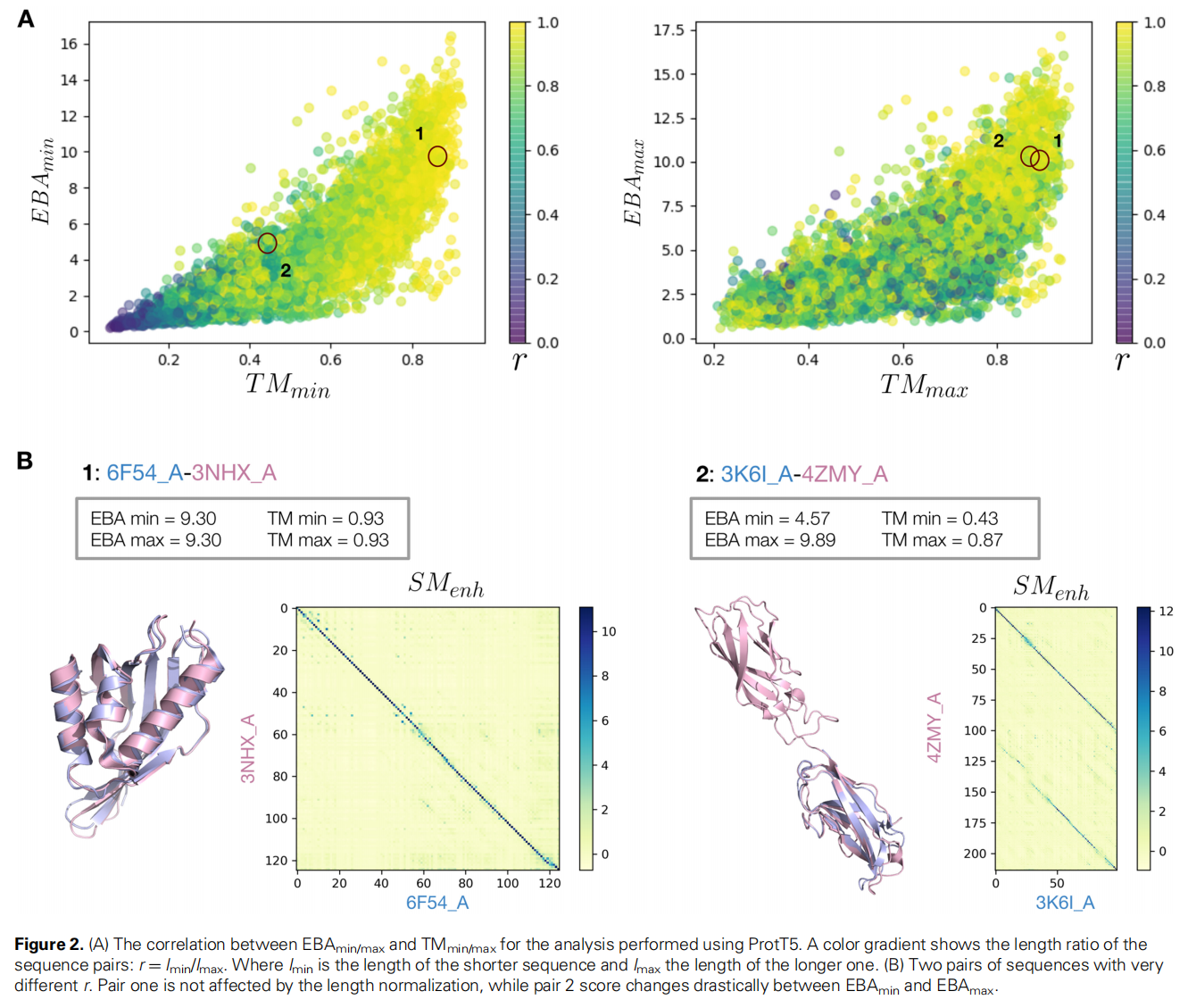
EBA successfully transfers CATH annotations
EBA competes with structure-based methods
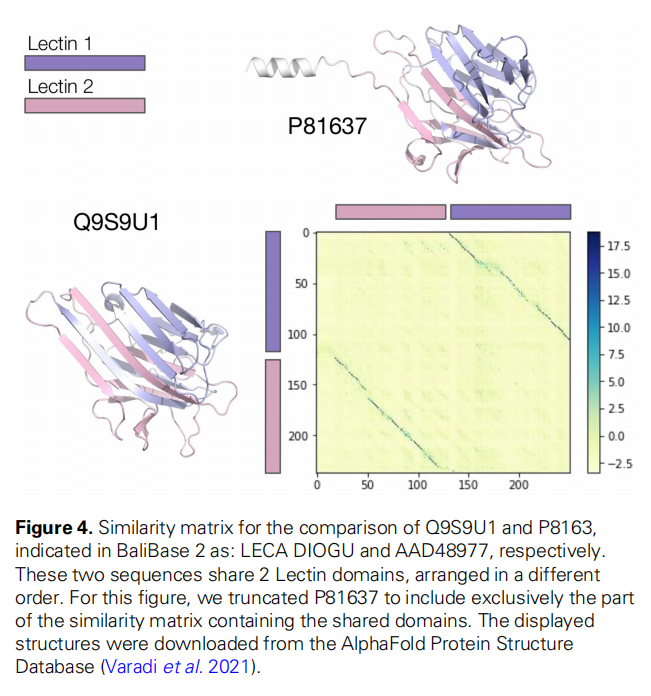
Domain permutation detection