论文实现:https://github.com/seanrjohnson/hmmer3di and https://github.com/seanrjohnson/esmologs
ESM-3B-3Di:ESM2+1D CNN预测3Di做同源检测
Abstract
基于profile和结构的方法在扩展到twilight zone of sequence相似度的时候需要很慢的预处理步骤,近年来使用深度学习方法用整个蛋白质和按位编码展现出了捕获长距离同源性的能力。本文展示了使用低维的位置嵌入能够直接实现速度优化的local search algorithms,使用ESM 3B将一级结构序列转化为3Di词表,并将这作为优化的Foldseek,HMMER3和HH-suite search algorithms的输入
Introduction
给一个没有实验的蛋白分配一个假设功能往往是通过找到在序列、结构或者进化上有关系的实验蛋白,将这个注释迁移过去,常见的方法有BLASTP,PSI-BLAST,HMMER3,HH-suite3和MMseqs2
sequence profiles来源于MSA,并且通过用hidden Markov models (HMMs)建模,比如HMMER和HH-suite,建模每个氨基酸上的概率和其插入或删除的概率,因为这种方法依赖于数据库建立,检索预处理所以开销较大
结构检索方法比序列检索更加的敏感,随着AlphaFold2和ESMFold等方法的进步与蛋白结构数据库的增长,一些快速的结构检索方法开发出来了,比如Foldseek,RUPEE,Dali
Results
Using ESM-2 3B to generate small positional embeddings
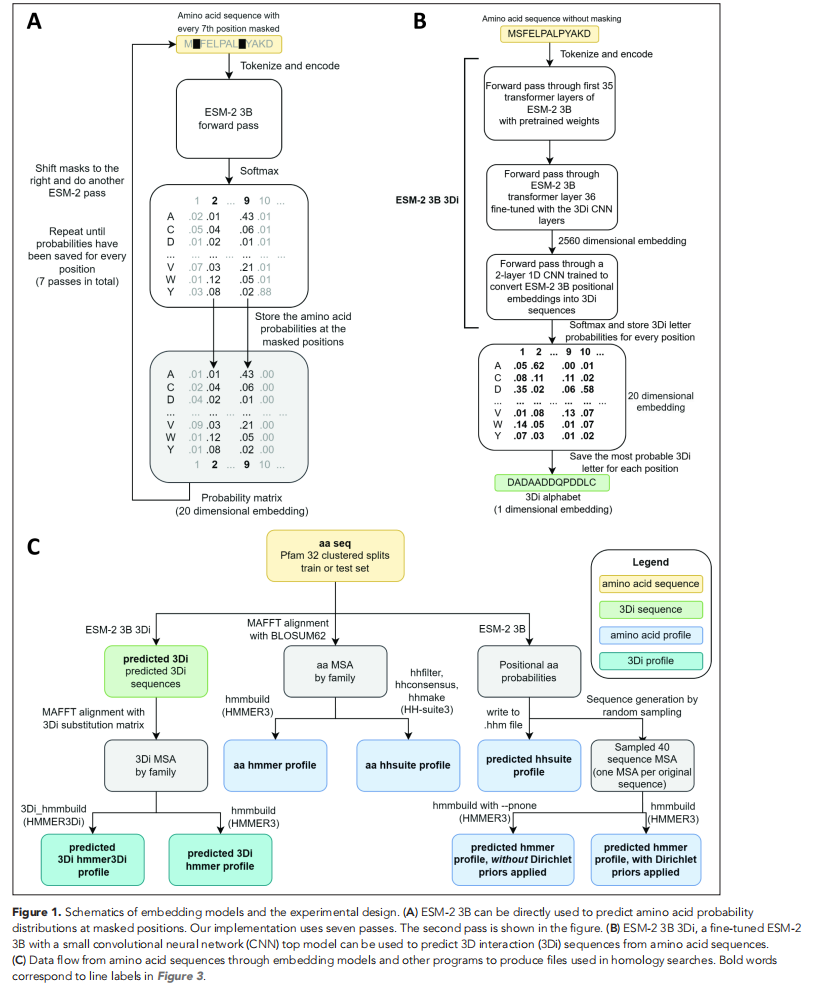
如figure 1A里面,生成概率给HMM tools;训练了一个额外的两层的1D CNN把ESM2 3B输入氨基酸序列转化为3Di,如figure 1B
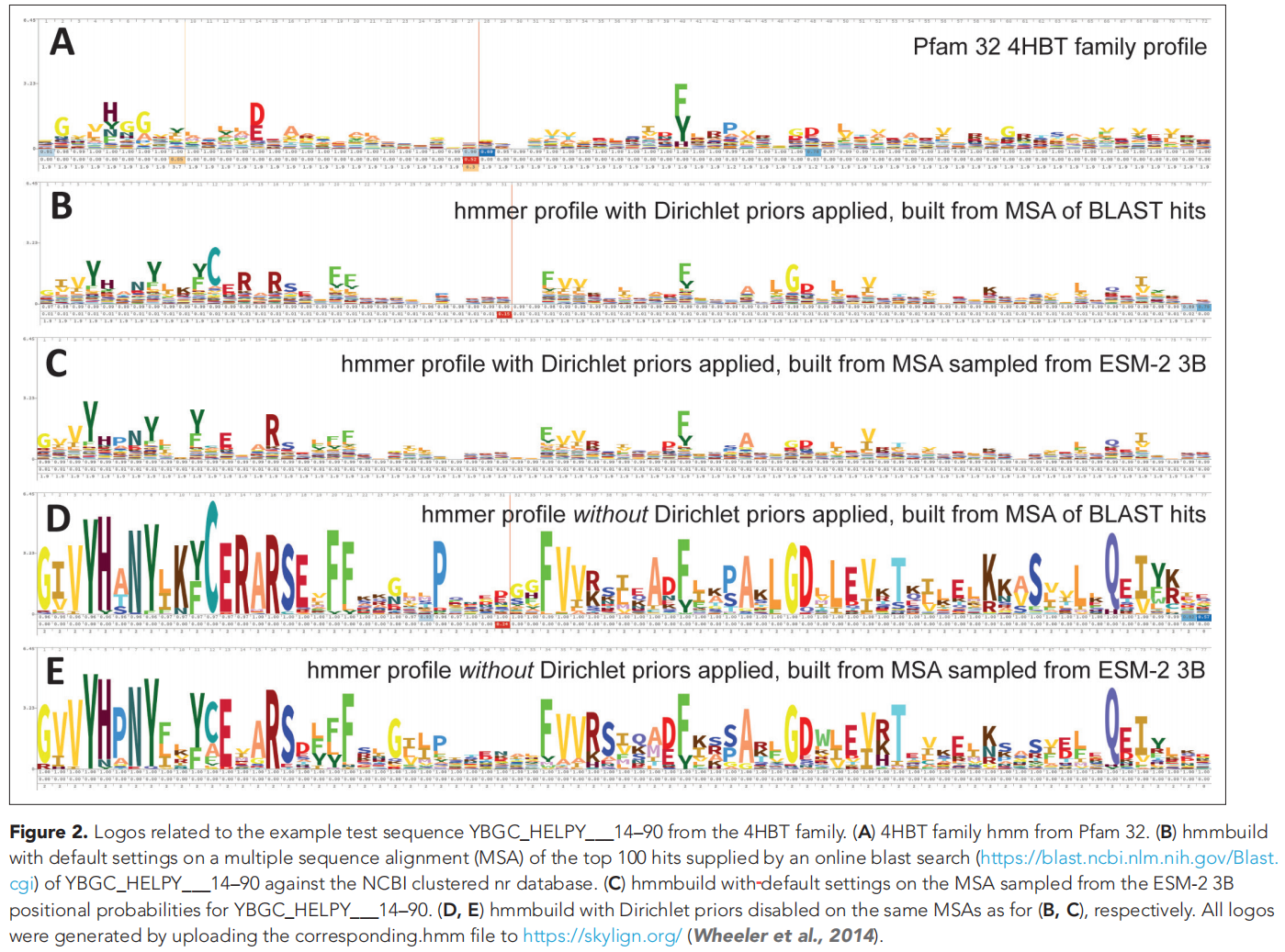
figure 2是用HMM做比对的一个例子
Comparison of newly developed embedding methods
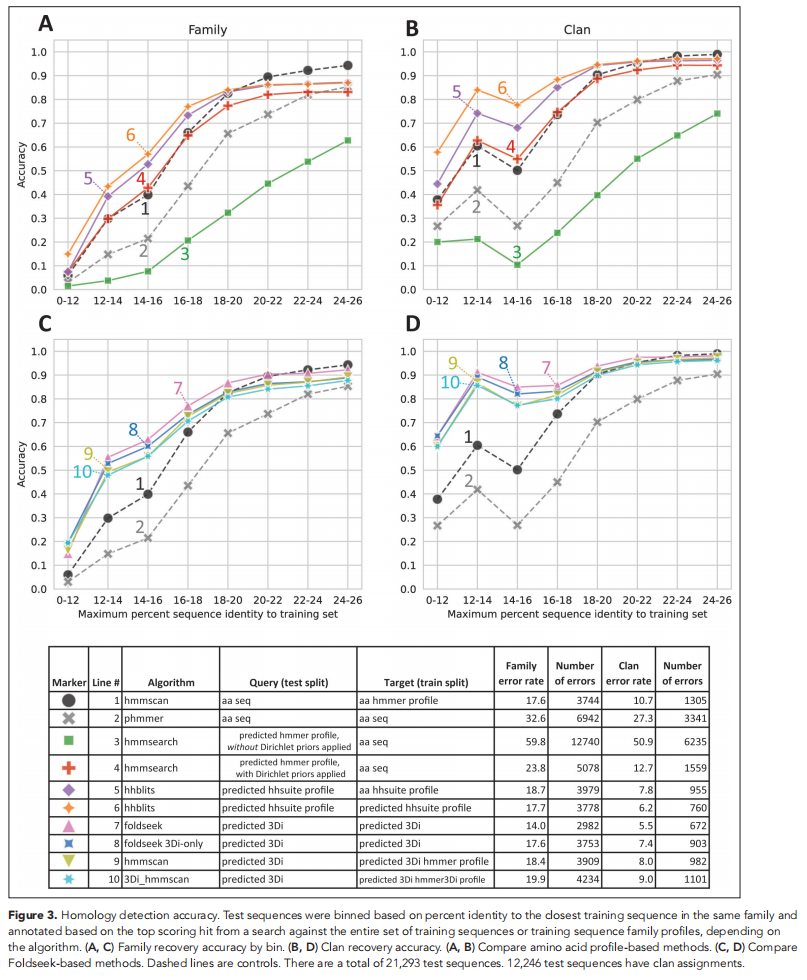
从Pfam 32 splits里生成predicted profiles和3Di sequence,然后转化为不同搜索工具的格式。Pfam中每个family划分的测试集保证了与训练集不超过25%的序列相似度
Foldseek搜索由ESM-2 3B 3Di figure 3 C,D在所有测试的新方法中表现最好
Benchmarking of ESM-2 3B 3Di against emerging and established search methods
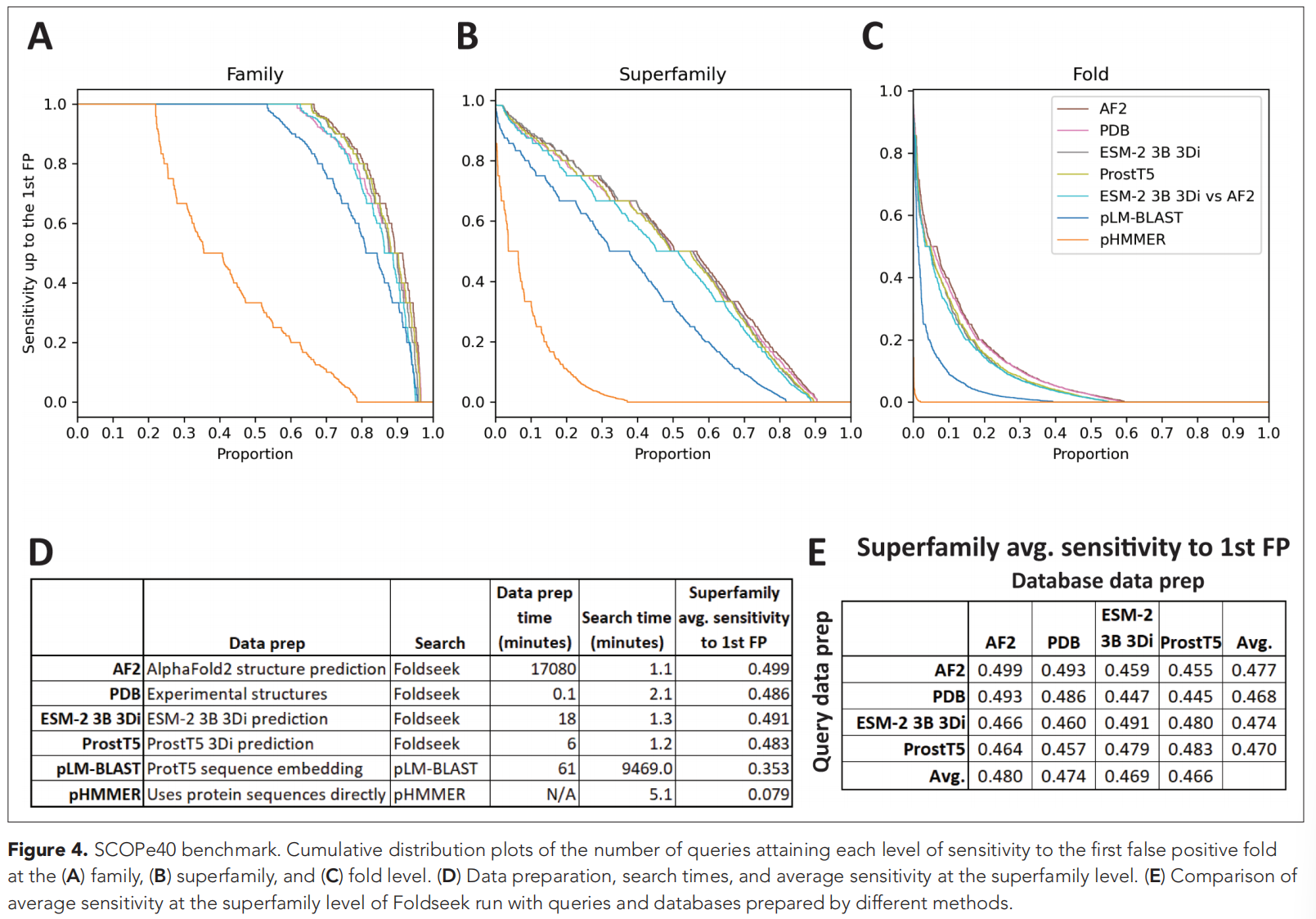
Discussion
比PLM-BLAST好,但是跟ProtsT5差不多
Methods
Alignments
MSA使用MAFFT (v7.505):–anysymbol --maxiterate 1000 --globalpair,BLOSUM62 substitution matrix, 3Di substitution matrix from Foldseek
Fine-tuning ESM-2 3B to convert amino acid sequences into 3Di sequences
训练集是Foldseek AlphaFold UniProt50 dataset 数据集划分(9:0.5:0.5)
Predicting profiles to generate HH-suite hhm files and HMMER3 hmm files from single sequences
- 因为ESM2预测第一个token倾向于预测M,但是pfam里面并不是,所以丢掉第一个
- mask第1、8…位置
- 得到每个mask的logits
- 把mask往右移一位
- 重复六次存下每个位置的logits

