ProteinGenerator:序列diffusion多场景蛋白质生成
Abstract
蛋白质去噪diffusion能够基于蛋白质骨架生成,但是在生成具有序列特异性和功能的蛋白上有限制,为了解决这个挑战提出了ProteinGenerator (PG),一个基于RoseTTAFold的序列空间diffusion能够同时生成序列和结构,从一个噪声序列表示开始,PG通过迭代去噪生成序列和结构对。设计了具有不同氨基酸组成和内部序列重复序列的耐热蛋白和笼型生物活性肽,如蜂黄蛋白。通过对具有不同结构约束的融合轨迹之间的序列logits进行平均,设计了多态父子蛋白质三组,当相同的序列完整或分裂成两个子域时,相同的序列折叠成不同的超二级结构。PG的设计轨迹可以由实验序列-活动数据来指导,为蛋白质功能的综合计算和实验优化提供了一种通用的方法。
Introduction
蛋白质功能起源于序列和结构之间的相互作用,因此设计新的蛋白质功能需要从序列和结构同时推理,大多数方法都是将结构和序列分开处理的,比如生成蛋白质骨架再通过逆折叠获得序列。传统方法比如Rosetta flexible backbone protein desgin是在序列和结构交替,而其他基于深度学习的方法都是先生成骨架,再序列设计比如ProteinMPNN。后者这类方法,DDPM在连续数据域中展现出相当大的前景。DDPM通过学习去噪被高斯噪声破坏的样本来近似于数据分布上的概率密度函数,能够从高斯先验中生成高质量的样本。尽管基于结构的方法比如RFdiffusion和Chroma非常有效,但在生成具有序列性质和识别多个fold或功能的序列上有所限制
作者推断,在序列空间而不是结构空间中进行扩散可以指导基于序列的特征和显式的序列设计。从RoseTTAFold结构预测网络开始,将输入序列和结构信息映射到输出序列和结构。RoseTTAFold可以通过标记PDB中的蛋白质序列来适应序列空间扩散,训练去噪的同时增加一个结构预测的loss来保证对序列和结构的理解
Results
Categorical DDPM implementation and fine-tuning

通过将蛋白质序列表示为离散的one-hot(对于原生序列,真值设置为1,所有其他值设置为−1)来实现扩散和数据噪声,噪音是高斯分布N(μ=0, σ=1)。为了微调RoseTTAFold,我们根据平方根计划输入逐步噪声的蛋白质序列(即噪声的强度与训练过程中的时间步相关联),输入数据不仅包括逐步加噪声的蛋白质序列,还可以包含可选的结构信息,用类别交叉损失来训练序列,用FAPE结构损失来训练结构。同时发现self-conditioning对于训练和推理的表现是有益的。
PG生成蛋白质从一个维度为L×20的高斯噪声序列开始,结构也被初始化。模型还可以结合序列指导信息,比如活性数据、序列特定的潜在信息或其他实验数据,将这些信息与模型当前的预测结果x0结合,引导模型朝着受限的序列空间(即符合特定要求的蛋白质序列)生成。有些motif(序列片段)是固定的,不能进行扩散处理,这些固定的序列位置通过额外的token来表示。二级结构信息(如α螺旋、β折叠等)通过一维轨道(1D track)传递,三维坐标信息(即蛋白质的空间结构)被嵌入到二维轨道(2D track)中,表示蛋白质中氨基酸对之间的距离或关系,以及三维轨道(3D track)则嵌入了实际的空间坐标信息,不同维度的嵌入信息(1D、2D和3D)通过跨注意力机制(cross-attention)在RoseTTAFold模型中进行结合
在推理阶段,模型从当前时间步的序列状态xt开始,预测出x0,即没有噪声的初始状态,然后,模型通过给x0加入噪声来生成下一个时间步的状态xt-1,这是一个逐步反向去噪的过程。同时比较两种采用方法:1. 从q(xt-1 | x0)进行采样,这意味着xt-1的生成仅基于去噪后得到的x0;2. 从q(xt-1 | x0, xt)进行采样,这意味着在生成xt-1时,模型会同时考虑去噪后的x0和当前的xt。并发现,第一种方式(仅基于x0进行采样)在实际操作中更加有效
PG容易设计出特定结构的motif,设计的与AF2的预测结构RMSD<2,motif RMSD < 1,AF2 pAE < 5。同时通过ESM pseudo-perplexity测量了序列质量,与UniProt24中采样的原生序列难以区分,并且明显高于使用6.4亿参数的序列扩散模型EvoDiff生成的序列
湿实验结果:
使用PG无条件生成,得到的序列-结构对的氨基酸组成类似于那些天然蛋白,figs7;生成的序列通过AF2和ESMFold折叠和设计的结构相似且可信。并无条件的生成了70-80氨基酸的蛋白质,通过体积排阻色谱(Size-Exclusion Chromatography, SEC)测试了溶解性和单体性,通过圆二色光谱(Circular Dichroism, CD)测试了折叠状态,通过CD热熔曲线(thermal melts)评估了热稳定性。
- 在42个经过实验测试的蛋白质中,有32个蛋白质在SEC测试中显示为可溶并且保持单体形式,表明这些蛋白质没有聚集成多聚体,并且在溶液中是稳定的。
- CD光谱实验表明,这些蛋白质具备设计的二级结构,即它们的折叠状态与设计时的预期一致。
- 热稳定性测试结果显示,这些蛋白质在高达95°C的温度下仍然保持稳定,表明它们具有良好的热稳定性,能够在高温下维持其结构不发生解折叠。
Design of rare amino acid enriched proteins
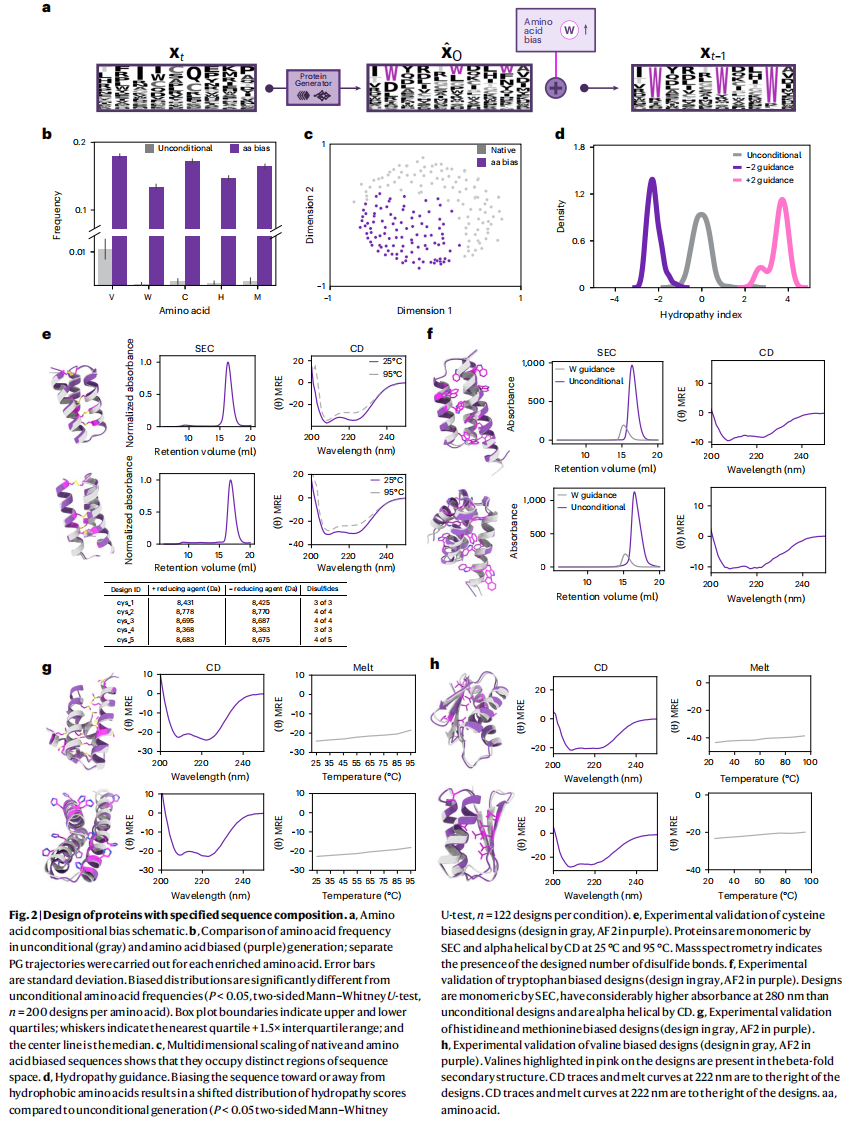
一个从序列空间上进行diffusion的优势是可以容易的实现基于序列的引导功能,能够在生成过程中通过特定的引导函数来影响生成的蛋白质序列,以满足某些特定的需求(如氨基酸组成)
为了评估PG模型在设计中是否能够超出其训练数据(来自PDB,即蛋白质数据银行)的分布,特别是在设计进化上较少见的氨基酸富集的蛋白质时,是否能够合理处理序列-结构关系,如fig2a
然后设计富含特定氨基酸的蛋白质,选择了一些在进化中较少出现的氨基酸,这些氨基酸具有重要的结构或功能特性,如色氨酸(Tryptophan)、半胱氨酸(Cysteine)、缬氨酸(Valine)、组氨酸(Histidine)和蛋氨酸(Methionine),根据这些目标氨基酸的频率对序列位置进行排序。
- 对排名前N个位置(其中N是目标氨基酸出现的期望次数),在生成下一时间步的序列xt-1时,会加入一个偏向这些氨基酸的引导,使这些位置更有可能包含目标氨基酸,生成了富含这些目标氨基酸(如色氨酸、半胱氨酸等,达到20%的组成比例)的蛋白质,fig2b
- 设计的蛋白经过过滤,确保它们具有较高的AF2预测可信度(pLDDT分数高于90)和自洽性(RMSD与设计结构小于2Å)
- 选择了96个蛋白质进行实验表征
实验结果显示,fig2e-fig2h:
-
68种在**大肠杆菌(Escherichia coli)**中成功表达并具有溶解性
-
在富含半胱氨酸的5个设计中,有4个在SEC(体积排阻色谱)实验中显示为单体;在富含色氨酸的19个设计中,有8个为单体;在富含缬氨酸的22个设计中,有19个为单体;在富含组氨酸的12个设计中,有10个为单体;所有富含蛋氨酸的10个设计都显示为单体;
-
富含半胱氨酸的蛋白质,fig2e:
- 在没有特定结构条件的情况下,富含半胱氨酸的设计生成了3-4个二硫键。这通过质谱实验在有无还原剂TCEP(50mM)的条件下进行了验证。二硫键的形成表明蛋白质中半胱氨酸之间发生了氧化还原反应,形成了稳定的结构。
-
富含色氨酸的蛋白质,fig2f:
- 这些蛋白质在280nm波长处具有较高的吸光度,这是色氨酸的典型特性。CD光谱显示这些蛋白质具有螺旋结构。
-
富含缬氨酸的蛋白质,fig2h:
- CD光谱显示富含缬氨酸的蛋白质具有较高的β折叠结构,这与缬氨酸的二级结构倾向相一致。此外,这些蛋白质在热稳定性测试中表现良好。
进一步探索了具有预先指定的电荷组成、等电点和疏水性的蛋白质的生成,fig2d
Design of sequence repeat proteins
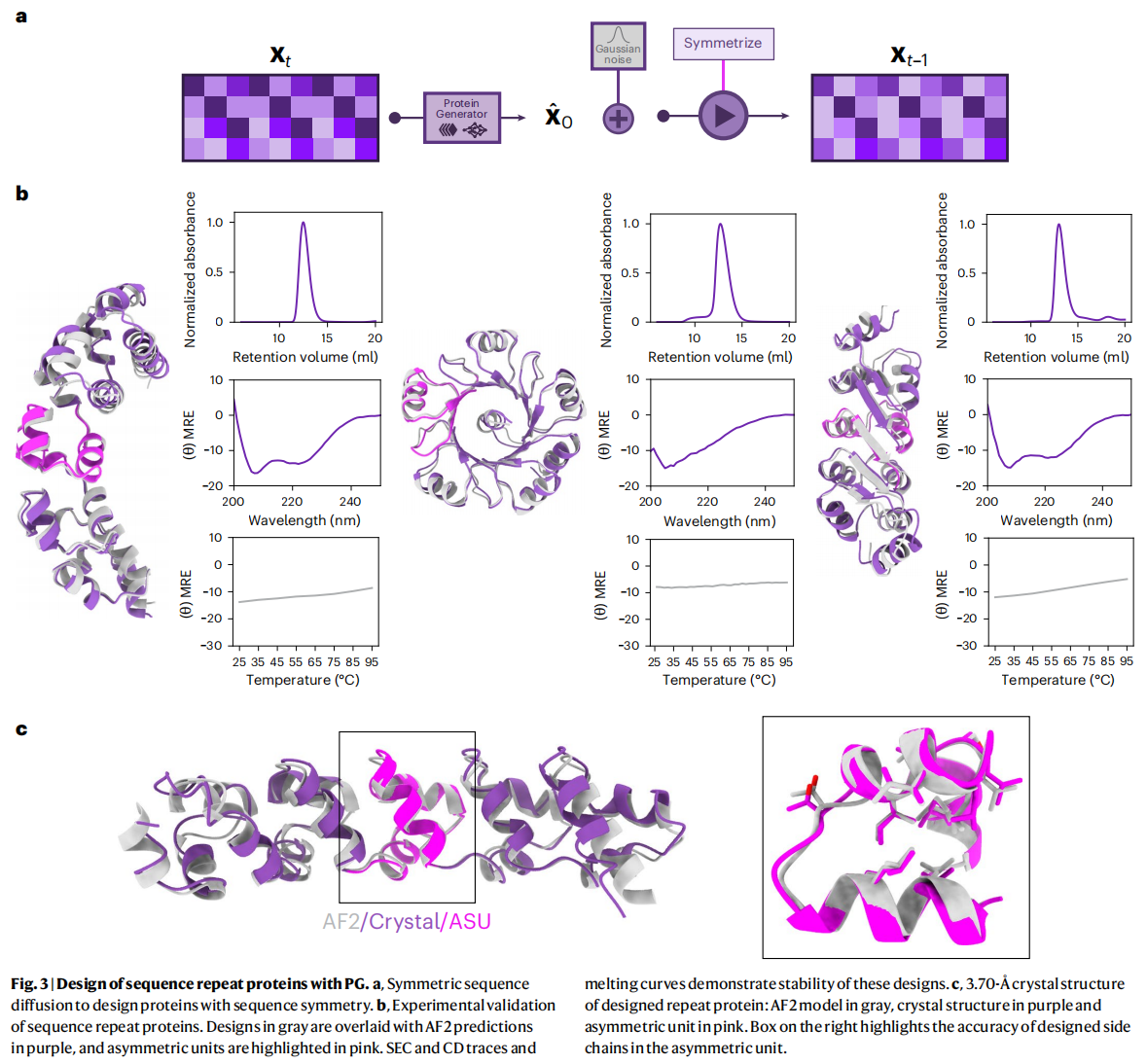
重复蛋白是指包含多个重复的序列-结构单元的蛋白质,在自然界中非常普遍。这类蛋白质在分子识别和信号传导等生物功能中起着重要作用。
PG模型的设计思路是,给定重复单元的序列长度和所需的重复次数,模型可以在每个时间步中应用**重复对称性(repeat symmetry)**来对加噪的序列分布进行处理。通过这种方式,PG模型能够生成带有重复单元的序列,并逐步去噪,最终生成符合设计要求的重复蛋白,fig3a
在没有特定结构指导的条件下,PG模型生成的重复蛋白主要是β螺旋结构(beta-solenoid structures)。为了鼓励模型生成更多样化的蛋白质结构,对PG模型进行训练,使其能够基于二级结构进行条件化生成。通过指定二级结构约束,模型可以生成多种类型的设计,包括全α螺旋、全β折叠,以及α螺旋和β折叠混合的结构,fig3b,为了提高设计蛋白的稳定性并减少聚集,研究人员在部分设计中加入了螺旋帽(helical caps)
湿实验结果:
实验验证了74个带螺旋帽的重复蛋白和86个不带螺旋帽的重复蛋白
- 在这160个重复蛋白中,27个带螺旋帽的设计和10个不带螺旋帽的设计在体积排阻色谱(SEC)测试中显示为可溶且为单体,表明这些蛋白质在溶液中没有发生聚集,且以单体形式存在
- 使用圆二色光谱(CD)对其中的8个蛋白质进行了测试,结果显示7个蛋白质具有预期的二级结构,验证了PG模型生成的蛋白质结构符合设计目标
对一个由五个重复单元组成的设计蛋白质进行了晶体结构解析。这个设计是由一个四螺旋束(four-helix bundle)的不对称单元构成,晶体结构解析显示,该设计具有原子级精度,即生成的蛋白质结构与晶体结构非常接近,fig3c
Design of conditionally active peptide cages for membrane lysis
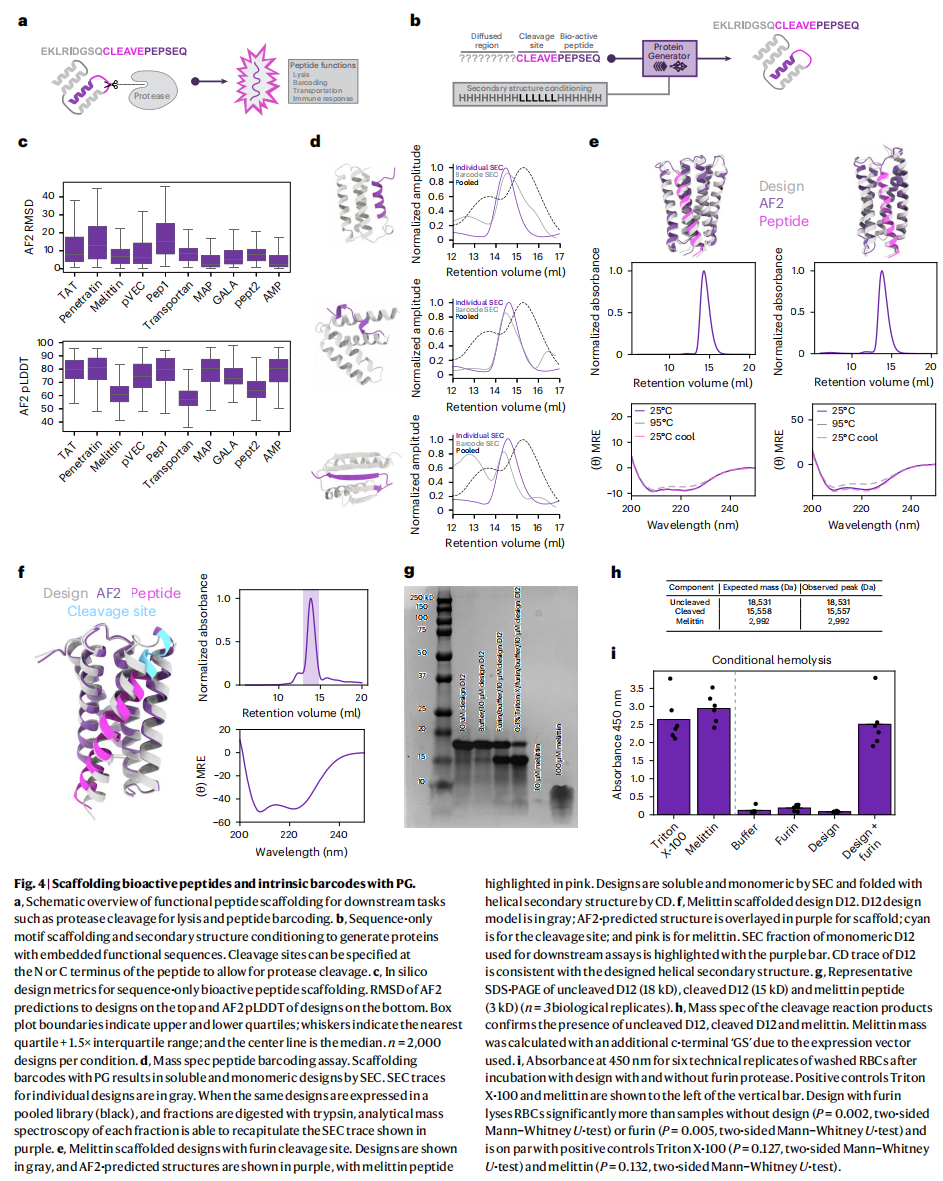
条件性活性意味着蛋白质的功能或活动是由外部输入(例如特定的信号、化学物质或环境变化)触发的。这种设计在治疗药物和生物传感器中非常重要,因为它们需要根据环境或外部刺激进行精确控制,以便在特定时间或空间内发挥作用。例如,在治疗药物中,蛋白质可以根据病情或特定体内信号激活;而在生物传感器中,蛋白质可以对某种分子或物理信号产生响应,fig4a
使用PG模型设计了包含生物活性肽序列的蛋白质。具体方法是将活性肽序列嵌入到一个惰性蛋白笼中(惰性蛋白质指的是没有活性的、结构稳定的蛋白质),这样肽序列可以保持其功能活性,而蛋白笼则提供稳定的支架和保护。指定了蛋白质链的某一区域(通常是N端或C端)为固定的生物活性肽序列。这意味着该区域的序列在生成过程中不会扩散或改变,从而保持其生物活性,其余的蛋白质序列则进行自由扩散,由PG模型生成,fig4b
PG生成的设计不仅能够成功整合肽序列,还能预测该蛋白质在三维空间中正确折叠,具有较高的可信度(pLDDT大于85)和与AF2精度(与设计的蛋白质笼的RMSD小于2Å)
使用PG模型设计一种蛋白质,这种蛋白质能够封闭(cage)一种名为melittin的生物活性肽,并在特定条件下释放它,尤其是在蛋白酶切割时释放,fig2b
melittin:
- Melittin是一种孔形成肽,来自蜂毒,具有强烈的细胞破坏作用,因为它能够在细胞膜上形成孔,导致细胞内容物泄漏。因此,melittin必须被严格控制,不能随意暴露,否则可能会导致细胞损伤。
- 研设计了一种蛋白质笼,将这种具有破坏性的melittin肽封闭起来,使其在特定条件下才会释放。
条件性释放:
- 研究人员引入了一种条件性释放机制,即通过蛋白酶切割来释放melittin。
- 为了实现这一点,他们在melittin的旁边设计了一个furin切割位点。Furin是一种常见的蛋白酶,能够在特定的序列上切割。通过引入这个切割位点,研究人员可以控制melittin的释放:当furin切割蛋白质笼的一个末端环结构时,melittin就会被释放出来。
二级结构引导:
- 为了保证melittin的功能正常,同时便于蛋白酶接触到切割位点,研究人员使用**二级结构引导(secondary structure conditioning)**来设计蛋白质笼。
- 他们将melittin嵌入到一个**螺旋束(helical bundle)结构中,并将切割位点(furin切割位点)放置在一个环结构(loop)**中,以便蛋白酶更容易接触和切割这个位点。这种设计增加了蛋白酶对切割位点的可接近性,从而提高了melittin的可控释放性。
多重约束:
-
在设计过程中,研究人员面临多重约束,主要包括:
- 将melittin序列嵌入到一个有序的结构中(如螺旋束),确保其在结构中能够正常发挥功能。
- 将furin切割序列置于环结构中,确保蛋白酶能够容易地切割这一部分以释放melittin。
melittin的无序性:
- Melittin肽在单独存在时通常是无序的,也就是说,它在天然状态下不会自动形成特定的结构。这给设计带来了挑战,因为研究人员希望通过模型使其在封闭结构中形成螺旋结构。
- 尽管melittin在孤立状态下是无序的,PG模型还是能够生成使melittin序列采取螺旋结构的解决方案。
实验验证,fig4e,f:
-
研究人员对13个通过PG模型生成的设计进行了实验验证,结果显示:
- 5个设计在实验中表现良好,表现为可溶且为单分散(即蛋白质在溶液中保持单体状态,没有形成聚集体),通过体积排阻色谱(SEC)进行了验证。
- 这些蛋白质在圆二色光谱(CD)测试中显示出具有螺旋二级结构,表明PG模型生成的设计能够成功地使melittin序列折叠成螺旋结构。
- 这些蛋白质在热稳定性测试中也表现良好,说明它们在高温条件下仍能保持其折叠结构,不会解折叠。
fig4h,这个表格展示了通过质谱分析确认的不同蛋白质的质量与预期质量的对比。未切割蛋白质(Uncleaved)的质量为18,531 Da,与预期相符。切割后的蛋白质(Cleaved)的质量为15,558 Da,表明切割后蛋白质损失了部分结构(即melittin被释放)。释放的melittin肽的质量为2,992 Da,也与预期一致,表明melittin肽在切割后被完整释放。
fig4i,条件性红细胞溶血实验展示了设计的封闭melittin蛋白质在特定条件下是否能够破坏红细胞膜。Triton X-100是阳性对照,显示了最高的溶血效果(强吸光度)。游离的melittin肽(第二个柱子)也表现出高溶血活性,表明melittin能够破坏细胞膜。未处理的缓冲液和furin蛋白酶本身(第三、四个柱子)没有明显溶血作用,作为阴性对照。
Scaffolding barcode peptide sequences
肽条形码(Peptide barcoding)是一种技术,用于在大规模蛋白质库中进行筛选。例如,在结合实验或体积排阻色谱(SEC)中,通过使用条形码可以标识每个蛋白质的身份。条形码的身份可以通过质谱检测出来(例如在蛋白酶切割后读取条形码肽的序列)。
当前的方法通常通过在蛋白质的N末端或C末端附加一个灵活的条形码序列,但这种方式可能会影响蛋白质的表达和溶解性,使得实验结果不准确。为了解释无条形码蛋白质的行为,通常需要使用多个(5-10个)变体条形码来推断其行为。
设计了一个带有条形码的蛋白质结构,将条形码嵌入到蛋白质的C末端,并且在条形码的两侧添加了赖氨酸(Lys)和精氨酸(Arg),这样可以通过Lys-C蛋白酶和胰蛋白酶进行切割,方便释放条形码。
实验验证:
创建了一个由84个设计蛋白质组成的初步条形码蛋白质库,并通过SEC分离这些蛋白质,fig4d
- 研究人员通过SEC分离这些设计蛋白质,并利用质谱(Orbitrap Lumos)检测各个分段中的条形码,识别每个蛋白质的身份。
- 作为对照,研究人员分别表达和纯化了这84个蛋白质,结果显示其中64个蛋白质成功表达,并且在这64个蛋白质中有48个蛋白质表现出预期的单分散洗脱峰,即这些蛋白质在溶液中保持单体状态。
- 他们将这些设计蛋白质的SEC洗脱峰与质谱重构的条形码对应的洗脱曲线进行了对比,结果显示73/84的设计在条形码实验中表现良好,其中58个设计的蛋白质通过条形码验证,其中41个设计(71%)的SEC洗脱体积与条形码检测的质谱信号一致。
Multistate design
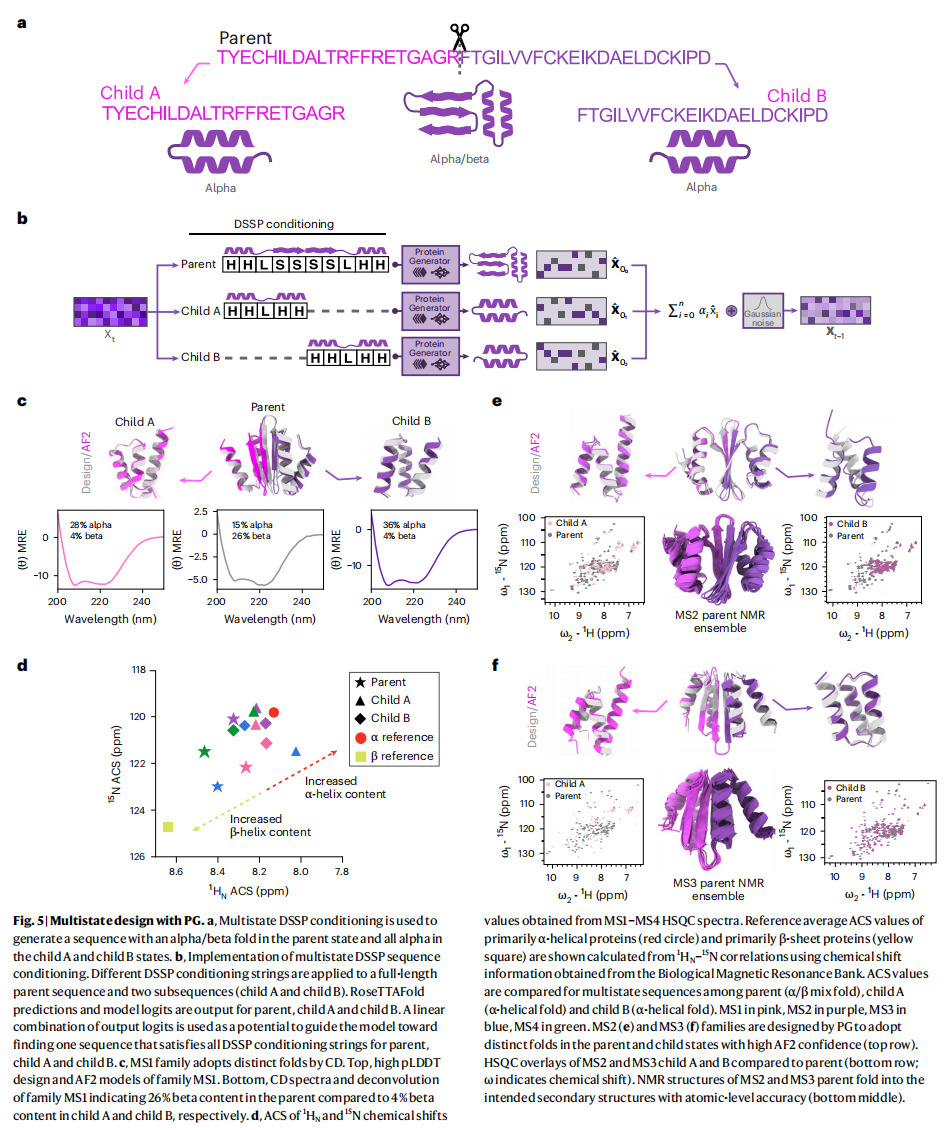
多态设计指的是设计一种氨基酸序列,使其在不同的条件下能够采用不同的结构构象。也就是说,同一个序列在某种外部刺激下(如切割、化学信号等)能够从一种结构转变为另一种结构。这种设计具有挑战性,因为蛋白质的能量景观必须包含两个或多个离散的能量最低点(即两个不同的结构状态),并且这两个状态之间的自由能差异必须足够小,才能让外部触发机制引发这种状态的切换。
输入给RoseTTAFold的并不是不同的序列,而是相同的序列但带有不同的结构调控信息(structural conditioning information)。换句话说,PG模型通过为同一个序列提供不同的结构约束条件,让模型可以生成多个不同的结构状态。在每个时间步的扩散过程中,模型根据这些不同的结构信息产生不同的logits(预测输出),然后对这些logits进行线性组合,作为输入传递到下一时间步
希望用PG模型设计出一种蛋白质序列,当以单条链的形式连接时(即父状态),它会折叠成一种结构;而当这条链被蛋白酶切割成两条链时(即子状态A和B),它们会折叠成不同的结构。这种设计允许一个序列在不同状态下采取不同的折叠方式,fig5a
- 在每个时间步(xt),研究人员使用RoseTTAFold模型来模拟全长父序列及其切割后的子序列(子状态A和B)。
- 模型会对父序列和两个子序列分别进行预测,并得到每个序列的logits(预测输出的概率分布)。这些logits随后被平均,再加入噪声生成下一时间步的序列(xt-1)。
DSSP特征被附加到每个家族成员的L×20序列表示中,以使蛋白质二级结构的条件作用,这些特征有助于模型在设计过程中对二级结构进行调控,使模型能够生成在父状态下采用α/β折叠,而在子状态下则采用全α螺旋折叠的多态序列,fig5b
研究人员首先对72个父-子三联体设计进行了实验验证,确认这些蛋白质在未切割时处于父状态,在切割后进入子状态,AlphaFold2(AF2)预测的蛋白质结构具有高度的可信度和准确性。进一步选取了4个可溶且单分散的序列家族(标记为MS1–MS4)进行更详细的研究,这些家族的父-子结构在CD(圆二色光谱)和核磁共振(NMR)实验中进行了表征,fig5c,d,e,f
Guidance with experimental data
Protein Generator (PG) 模型结合实验数据来优化蛋白质功能,特别是在定向进化(directed evolution)过程中
使用了一组针对GB1蛋白(IgG结合蛋白)的实验基准数据集进行测试。这个数据集的优势在于其完整性,即所有可能的四个氨基酸位点的组合及其对应的活性数据都已知。因此,研究人员可以直接评估PG模型生成的每个序列的活性。
- 通过前几轮实验确定的适应性数据训练分类器(如多层感知器,MLP),并使用这些分类器的梯度来引导模型生成序列的采样过程。
- 在每轮实验中,研究人员生成了96个设计并进行了测试,总共进行了三轮。
- PG模型生成的设计在每一轮实验中平均和最大适应性都有所提高,且表现优于贝叶斯优化的基线模型
Discussion
PG模型能够生成多种不同类型的蛋白质,满足不同的序列域约束,包括氨基酸组成偏差、重复序列对称性、生物活性肽的封闭设计以及多态设计。这些功能展示了PG在处理复杂蛋白质设计任务中的灵活性。
优势:
- 多态设计是PG的一个特别优势,指的是蛋白质序列能够折叠成两种或更多的不同结构。比如,在前面提到的父-子三联体设计中,PG通过logit平均(对不同状态下的预测结果取平均值)实现了多态设计。这种方法在结构空间扩散中不容易实现,因此RFdiffusion不适合处理这种任务。
- PG模型能够在不依赖于固定结构先验的情况下,通过联合搜索序列和结构空间,进行深度学习驱动的多态设计。这种能力可以推广到更复杂的状态切换蛋白质系统的设计,这将有助于未来开发更复杂的条件性蛋白质系统(即蛋白质在外部刺激下切换状态的系统)。

