论文地址:http://biorxiv.org/lookup/doi/10.1101/2022.07.10.499510
DiffAb:扩散模型做序列-结构联合设计/给定结构的序列设计/抗体优化等
Abstract
抗体和抗原的结合主要取决于抗体的CDR区域,本文介绍了一个基于扩散概率模型和等变图神经网络的CDRs序列和结构生成模型,能够进行序列结构协同设计、给定主干结构的的序列设计和抗体优化等,在生物物理能量函数和其他蛋白质设计指标测量的结合亲和度方面产生有竞争的结果
Introduction
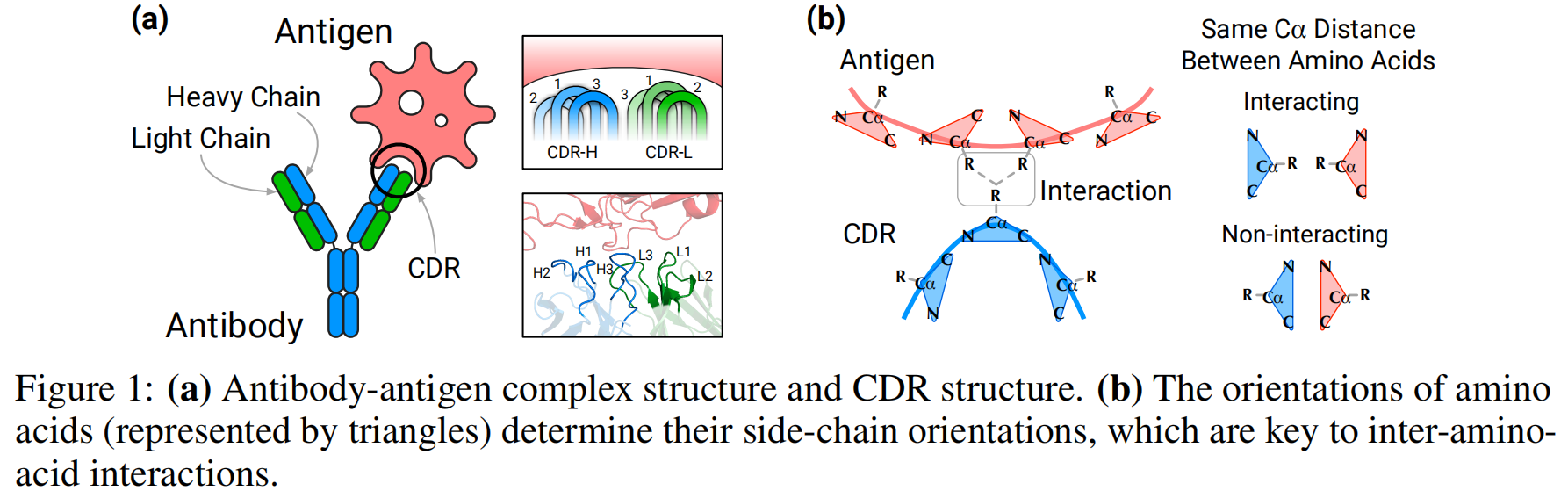
一种抗体包含两条重链和两条轻链,其整体结构相似。六个可变区域决定了一种抗体对抗原的特异性。它们被称为互补性决定区域(CDRs),记为H1、H2、H3、L1、L2和L3。
传统方法依赖于从复杂的生物物理能量函数中取样蛋白质序列和结构,通常是耗时的,而且很容易陷入局部最优状态
现有方法无法满足以下三个关键设计原则:
- 模型必须显式地基于抗原的 3D 结构来生成与之匹配的 CDR
- 模型需要同时考虑氨基酸的位置和方向(特别是侧链方向)来建模抗体-抗原的相互作用
- 除了从头设计,还应能够优化已有抗体以提高与抗原的结合亲和力
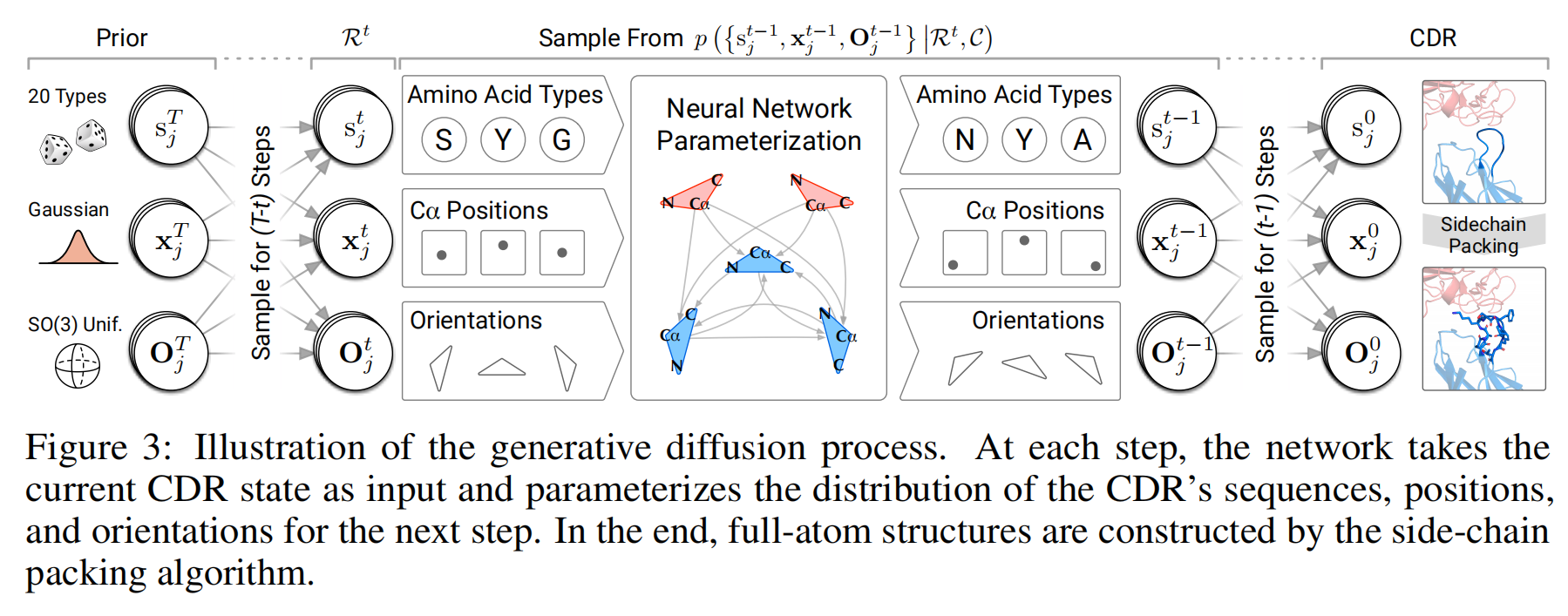
作者提出了一种基于扩散生成模型的框架,支持 CDR 的序列-结构联合生成,并直接条件化于抗原的 3D 结构。该模型首先聚集了来自抗原和抗体框架的信息。然后,迭代地更新CDR上每个氨基酸的氨基酸类型、位置和方向。在最后一步中,我们利用基于预测方向的侧链填充算法在原子水平上重建CDR结构。使用Diffusion的一个重要原因是它可以在序列结构空间中迭代地生成CDR候选对象,以便我们可以干扰并在采样过程上施加约束,以支持更广泛的设计任务
Methods
Definitions and Notations
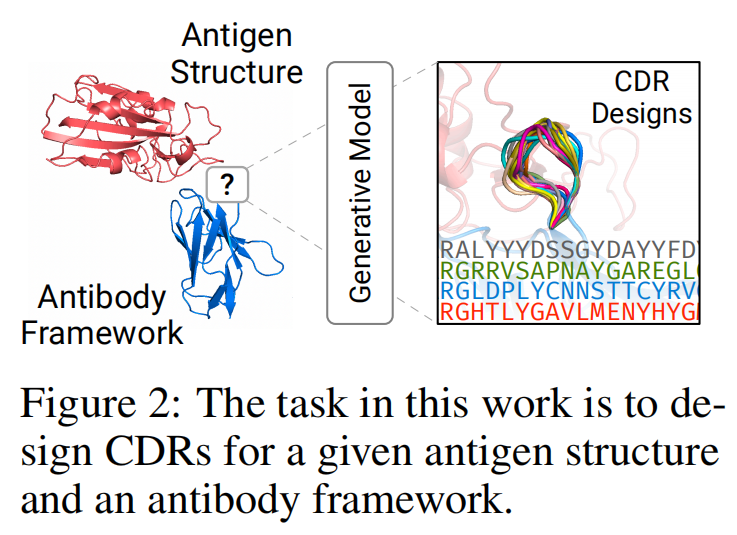
在这项工作中,假设给出了抗原结构和抗体框架(图2),重点是在抗体框架上设计CDR。氨基酸类型 $s_i$ ,主 $C_\alpha$ 坐标$x_i\in\mathbb{R}^3$,$O_i\in SO(3)$ 描述侧链方向的旋转矩阵
Diffusion Processes
Multinomial Diffusion for Amino Acid Types
单步扩散,在时间步t,氨基酸类型 $s_j^t$ 的分布由以下公式定义
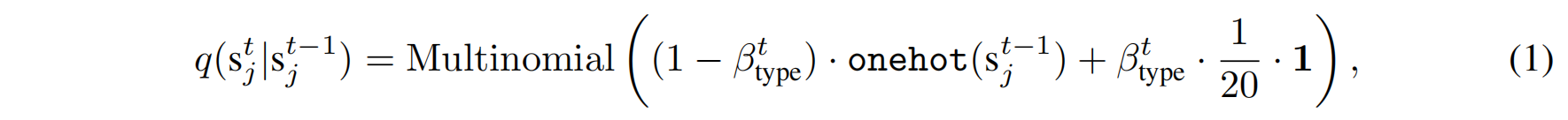
$\beta_t^{type}$ 控制噪音加入速率,随着时间步t增大逐渐增大
从初始数据扩散至任意时间步

当t -> T时, $\alpha_t^{type}$ -> 0,表示数据逐渐变成均匀分布
正向扩散过程不依赖于上下文或其他输入,仅根据噪声参数来平滑真实数据分布
生成扩散,在时间步t,氨基酸类型 $s_j^{t-1}$ 的生成分布定义为:
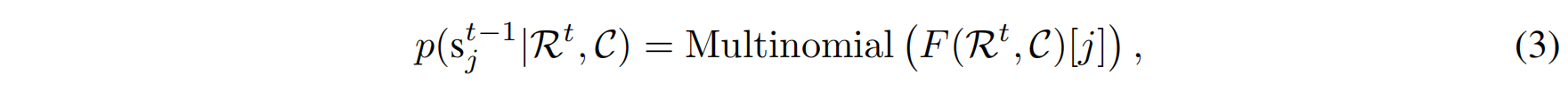
$F(R^t,C)[j]$ 通过神经网络预测的后验分布,条件化于:当前时间步的CDR状态和抗原抗体框架
模型需要最小化生成分布与真实后验分布之间的 KL 散度:
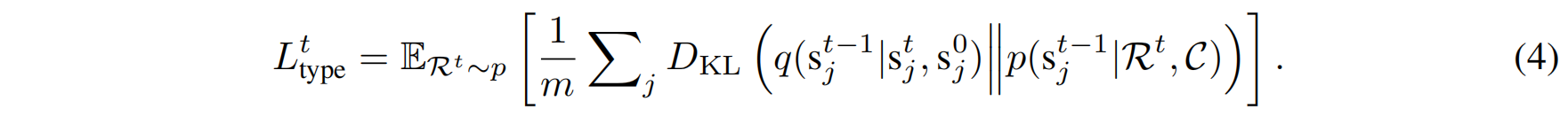
$q(s_j{t-1}|s_jt,s_j^0)$ 是正向扩散的真实后验分布, $p(s_j{t-1}|Rt,C)$ 是生成扩散的预测分布,m是CDR中氨基酸的数量
Diffusion for Cα Coordinates
在时间步t
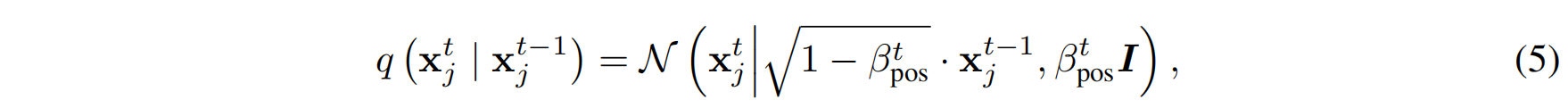
$\beta_t^{pos}$ 扩散速率0-1,随着时间步t增加逐渐增大,I单位矩阵,表示噪声的协方差矩阵,坐标数据逐渐平滑接近正态分布
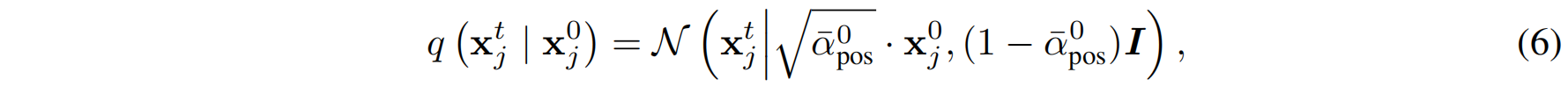
$\alpha_t^\mathrm{pos}=\prod_{\tau=1}t(1-\beta_\tau^\mathrm{pos})$ 表示当前分布与初始真实数据的相似程度
单步生成扩散
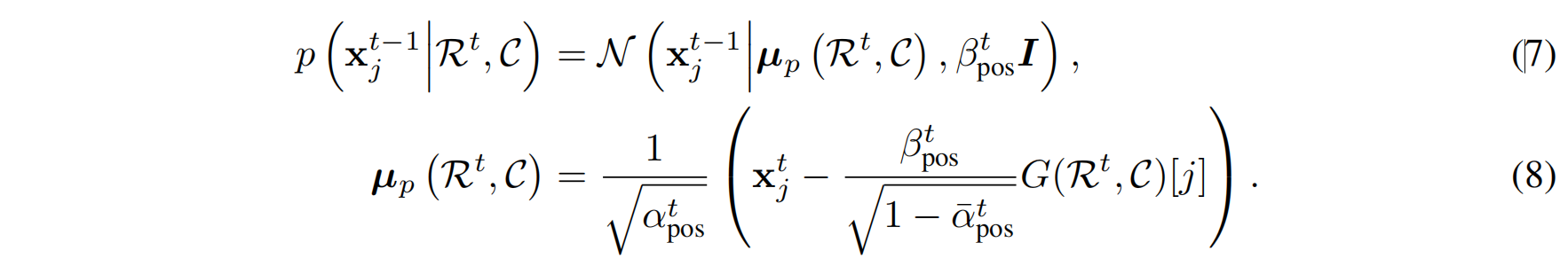
G是神经网络用于预测噪音
模型最小化生成分布与真实后验分布之间的均方误差 (MSE) 损失函数:
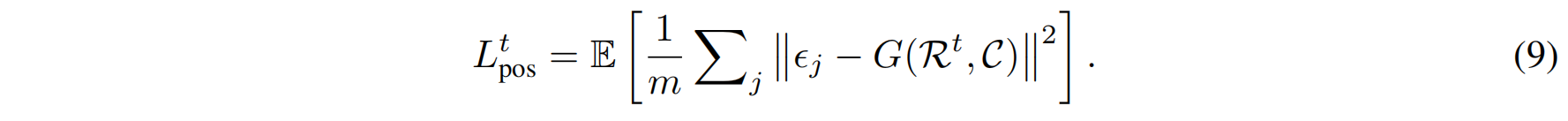
SO(3) Denoising for Amino Acid Orientations
利用 SO(3) 群(3D 空间中的特殊正交群)来建模氨基酸的旋转方向,能够确保生成的方向具有物理一致性和旋转不变性
单步扩散:

$\mathrm{IG_{SO(3)}}$ 是定义在SO(3) 群上的等变高斯分布, $\alpha_t^{ori}$ 是控制方向矩阵与初始真实方向的相似程度,随着时间步t增加而减少,ScaleRot 通过缩放旋转角度来模拟噪声的加入
当t->T, $\alpha_t^{ori}$ -> 0表示方向逐渐被完全随机化,接近均匀分布
生成扩散过程:
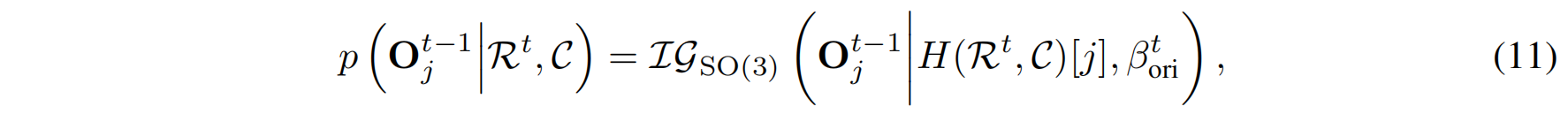
H是神经网络预测方向的后验分布, $\beta_t^{ori}$ 控制去噪过程中的不确定性
模型需要最小化生成分布与真实分布之间的距离,用 Frobenius 范数最小化真实方向和预测方向之间的误差
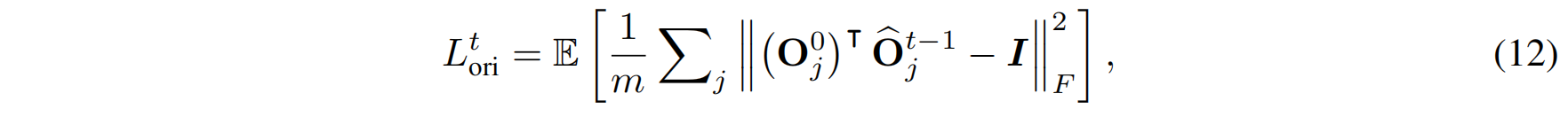
The Overall Training Objective
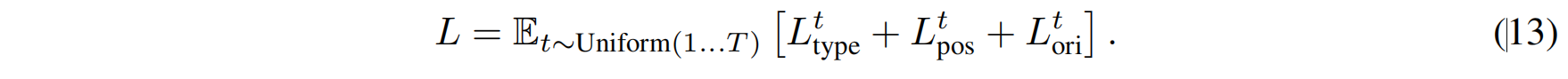
Parameterization with Neural Networks
$F(R^t,C)$ 用于预测氨基酸的类型(离散变量), $G(R^t,C)$ 用于预测氨基酸的坐标变化(连续变量), $H(R^t,C)$ 用于预测氨基酸的方向(旋转矩阵,SO(3))
每个神经网络的输入是当前时间步t和状态 $R^t$ (扩散模型生成的中间状态,包括氨基酸的类型、坐标和方向) 和上下文C (抗原和抗体框架的上下文,包括抗体骨架和抗原的 3D 结构)
-
定义等变性
对于任意旋转矩阵 $R\in SO(3)$ 和平移矩阵 $r\in \mathbb{R}^3$ ,网络满足
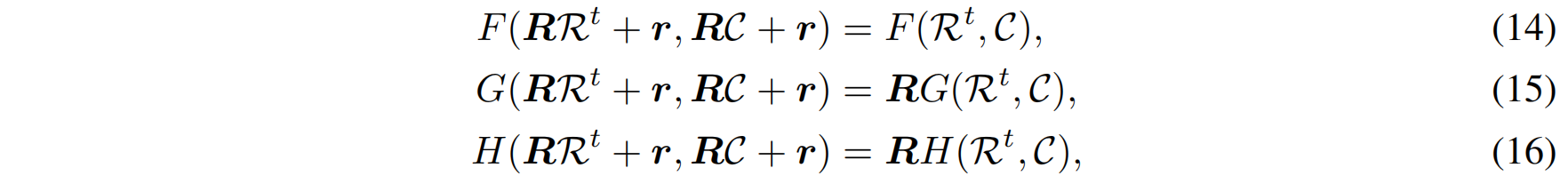
-
实现等变性
使用图神经网络或等变注意力模块,将氨基酸的几何关系嵌入模型,同时确保输出结果在旋转和平移下保持一致性
Sampling Algorithms
采样算法的核心是生成抗体的 CDR 序列和结构,分为以下三个步骤:
- 初始化:从均匀分布(氨基酸类型)或标准正态分布(坐标和方向)中随机采样初始状态。
- 逐步去噪:从时间步 T 开始,采样算法逐步逆向执行扩散过程,通过神经网络在每一步预测去噪值并更新状态
- 结构重建:在最终时间步 t=0 完成生成后,根据预测的坐标和方向用 侧链填充算法(如 Rosetta 的 packing 功能)补全侧链原子位置,生成完整的抗体结构。
Experiments
Sequence-Structure Co-design
数据清洗:数据来源于 SAbDab 数据库,移除分辨率差于 4Å 的结构,丢弃针对非蛋白抗原的抗体;
数据划分:根据 CDR-H3 序列(互补决定区)的 50% 序列相似性对抗体进行聚类,手动选择 5 个聚类作为测试集,总共包含 19 个抗体-抗原复合物,包含来自 SARS-CoV-2、MERS、流感等病原体的抗原
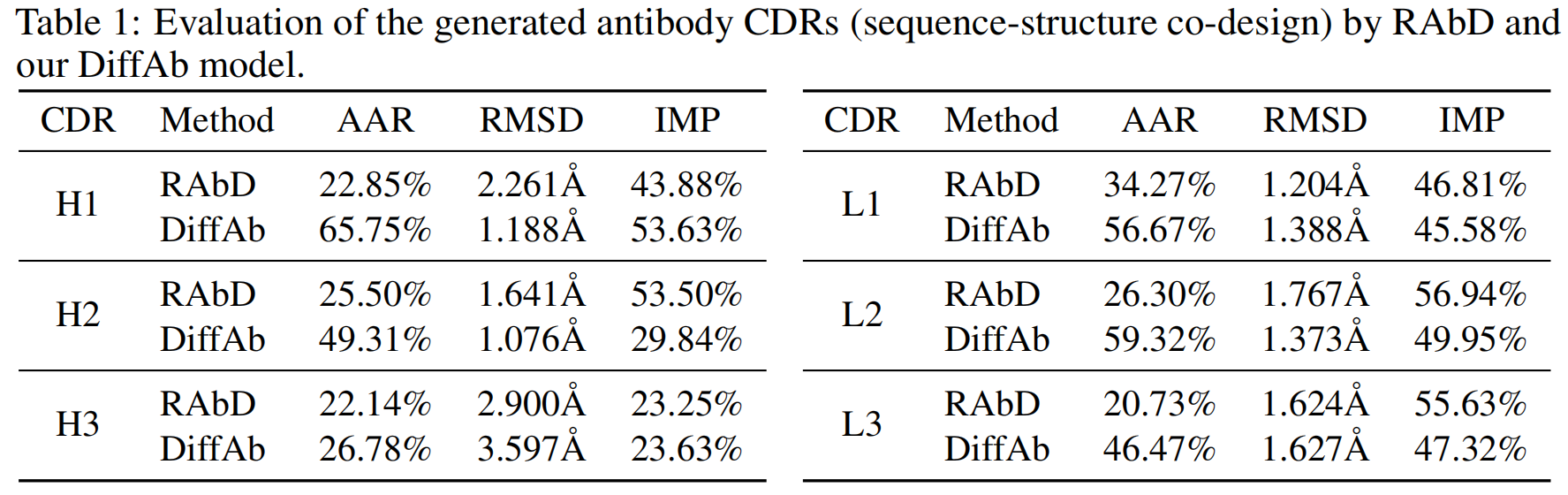
与 Rosetta 能量函数驱动的抗体设计软件 RAbD【Adolf-Bryfogle et al., 2018】进行比较,每种方法为每个 CDR 生成 100 个样本
指标:
-
Amino Acid Recovery (AAR):
- 定义:生成序列与参考序列的序列一致性(序列相似度)。
- 作用:衡量模型生成准确性。
-
Root-Mean-Square Deviation (RMSD):
- 定义:生成结构的 Cα 原子与原始结构的均方根偏差。
- 作用:衡量生成结构与真实结构的几何相似性。
-
Improved Binding Percentage (IMP):
- 定义:生成 CDR 与抗原的结合能低于原始 CDR 的比例(结合能由 Rosetta 的 InterfaceAnalyzer 计算)。
- 作用:评估生成抗体在结合性能上的改进。
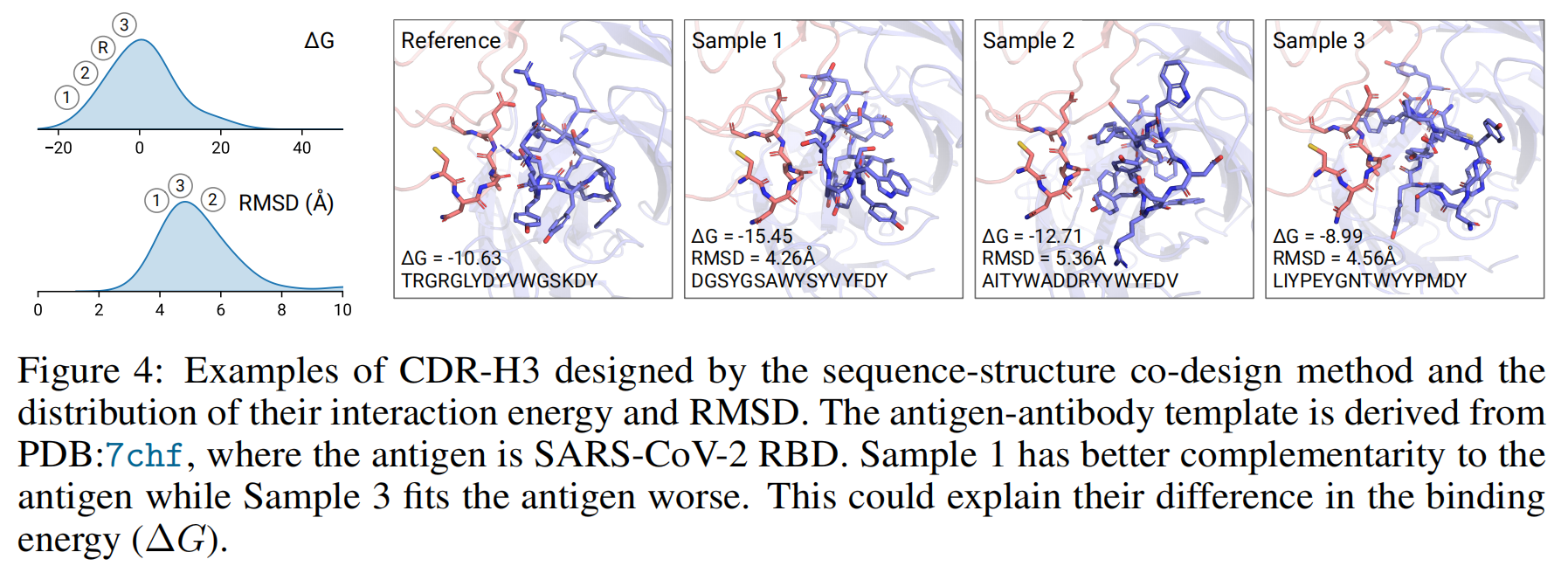
- 提供了针对 SARS-CoV-2 RBD(PDB: 7chf)生成的 CDR-H3 示例及其结合能量分布。
- 示例 1 与抗原的互补性最佳,结合能最低(ΔG = -15.45)。
- 示例 3 的形状不适合抗原,结合能高于原始抗体
Fix-Backbone Sequence Design and Structure Prediction
- 在此任务中,已知抗体互补决定区(CDR)的骨架结构,目标是基于给定的骨架设计出新的 CDR 序列。
- 同时模型可以通过固定序列来预测 CDR 的结构
基准方法使用 FixBB,这是一种基于 Rosetta 的序列设计软件,可以根据 CDR 骨架结构设计序列
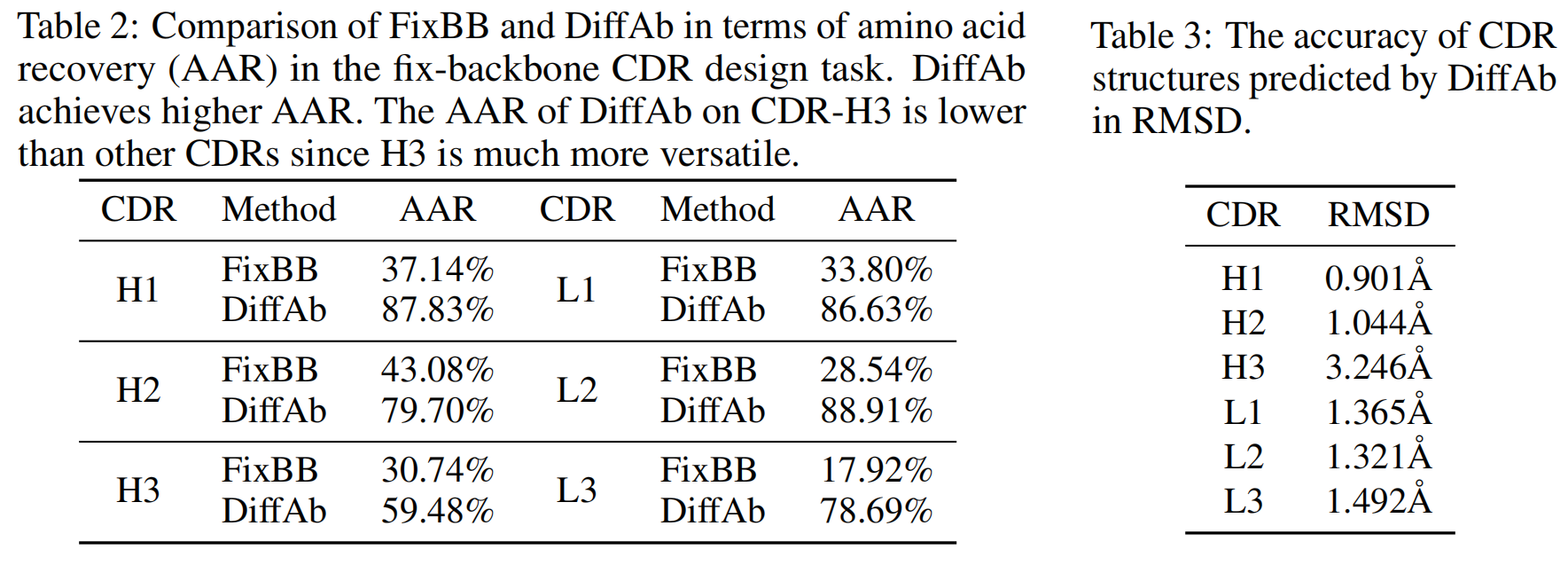
- 在 CDR H1、H2、L1、L2 和 L3 上,DiffAb 的结构预测精度非常高(RMSD ≤ 1.5 Å)。
- 在 CDR-H3 上,预测精度较低,主要由于其较高的变异性
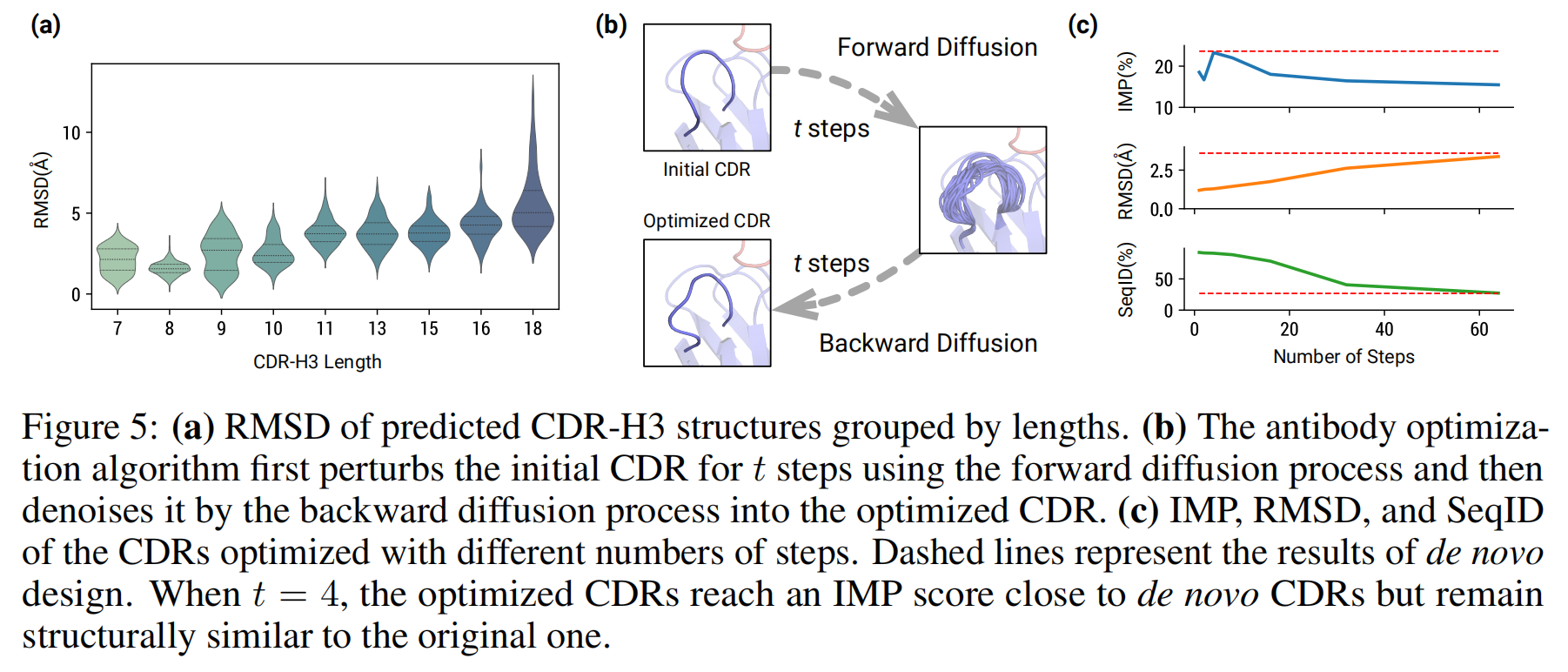
图5a 显示了不同长度的 CDR-H3 结构的预测精度,短序列的预测更准确,而长度超过10个氨基酸的 CDR-H3 预测精度降低
Antibody Optimization
对现有抗体的 CDR(互补决定区)序列和结构进行优化,以增强抗体与抗原的结合能力(使其与目标抗原的结合能降低),同时尽量保持原始抗体的结构和功能特性
先正向加噪:对原始抗体的 CDR 序列和结构进行 ttt 步的正向扩散(添加噪声),使其从原始状态逐渐接近随机分布;再从T-t开始反向去噪逐步生成优化后的CDR
对测试集中抗体的 CDR-H3 进行优化,每个抗体的优化重复 100 次,以生成 100 个候选的优化 CDR
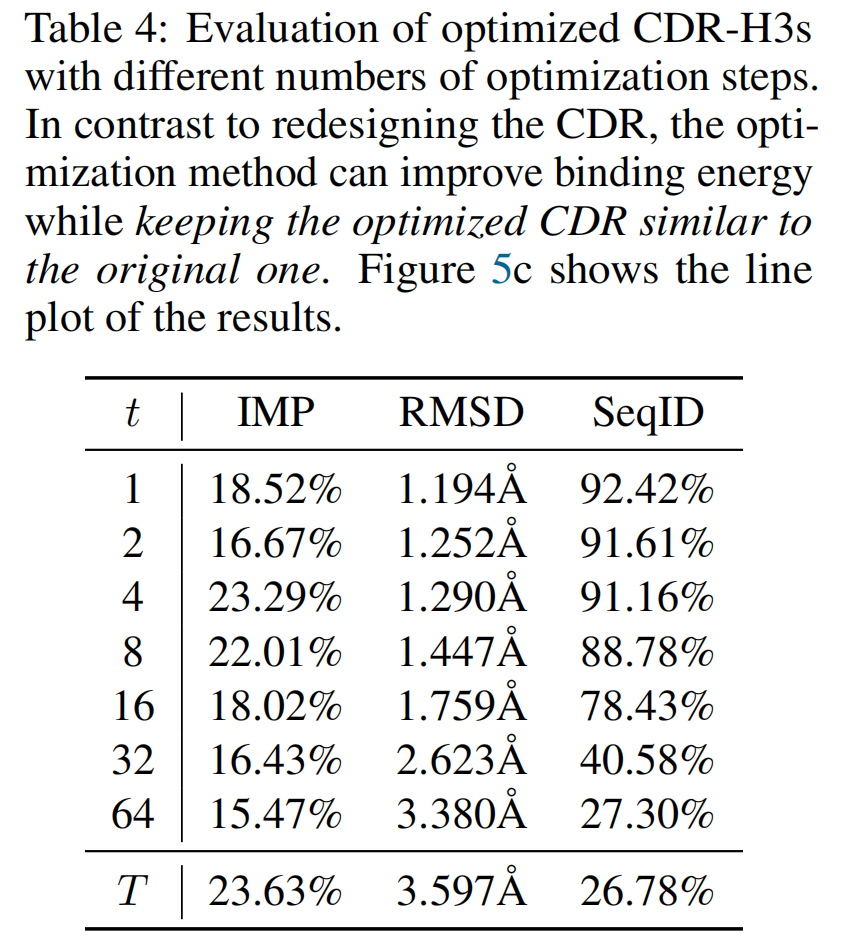
表4和图5c所示,通过 Rosetta 能量函数测量,优化方法可以产生抗体的结合能提高的抗体
-
结合能改进:
- 在较少优化步数(如 t=4)时,IMP 得分较高,说明优化后的 CDR 在结合能上有显著改进。
-
结构保持:
- RMSD 值随着 t 增加而升高,说明更多的优化步数导致结构偏离原始 CDR。
-
序列保持:
- SeqID 随着 t 增加显著降低,说明更大的步数会导致序列与原始序列的差异增大。
Design Without Bound Antibody Frameworks
在没有已绑定抗体框架的情况下,根据抗原信息独立设计 CDR,生成合理的序列和结构,模拟抗体-抗原的结合
-
输入条件:
- 抗原结构:目标抗原的 3D 结构(无已绑定抗体框架)。
- 抗体模板:通过 docking(分子对接)技术生成的抗体骨架模板。
-
对接(Docking):
- 使用抗体模板和抗原进行对接来生成初始抗体-抗原复合物。
- 对接工具:使用 HDOCK【Yan et al., 2017】进行对接。
-
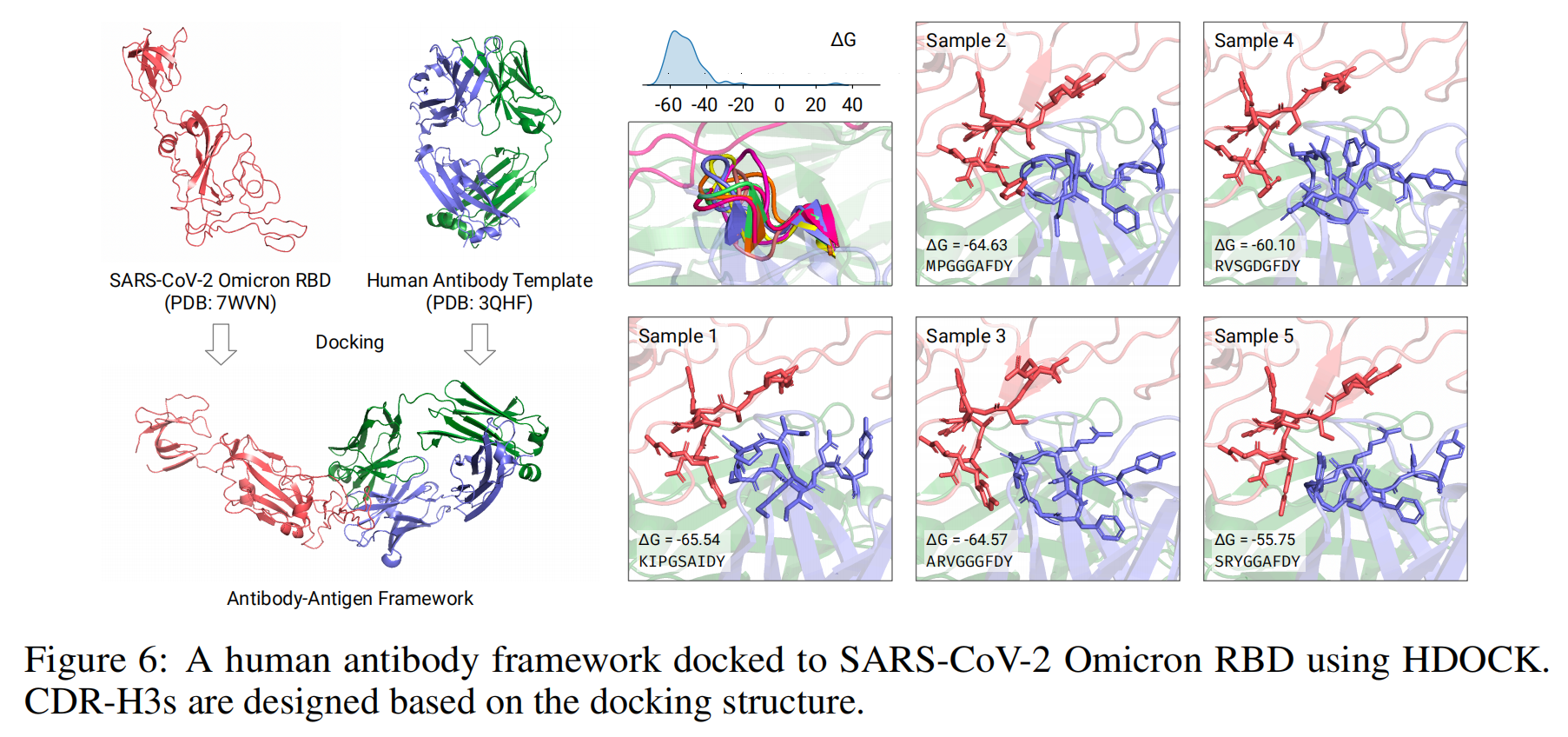
示例任务:
- 设计针对 SARS-CoV-2 Omicron RBD 的抗体。
- 抗体模板来源:针对流感病毒的抗体(PDB: 3bgf)。
- 初始 CDR-H3 的残基范围为 A322-A590
-
结构结果:
- 针对 SARS-CoV-2 Omicron RBD,生成的抗体在结构上表现出合理性,与抗原存在明显的结合位点。
- 图 6 显示了对接后的抗体-抗原复合物以及生成的 CDR-H3 的结合模式。
-
结合能结果:
- 生成的抗体虽然缺乏参考抗体的结合能力,但结合能分布表明其生成结果在合理范围内。
- 结合能(ΔG)可以作为生成抗体合理性的支持证据。

