论文地址:Conditional Antibody Design as 3D Equivariant Graph Translation
MEAN:内外编码器输入重链轻链抗原联合设计CDR序列和结构
Abstract
抗体设计在治疗用途和生物学研究中具有重要价值,但现有的深度学习方法存在以下问题:1. 对补体决定区(CDRs)的生成缺乏完整的上下文信息。2. 无法全面捕获输入结构的三维几何信息。3. 自回归生成方式效率低下,且易产生累积误差。本文提出MEAN模型,将抗体设计建模为一种条件图翻译任务,输入包括目标抗原和抗体轻链的信息,结合E(3)-等变消息传递和新型注意力机制,更好地捕获复杂几何关系,多轮渐进式全域解码同时生成CDR的1D序列和3D结构。MEAN在序列和结构建模、抗原结合的CDR设计以及结合亲和力优化任务中,显著优于当前最先进的方法,分别在抗原结合CDR设计和亲和力优化任务上提升约23%和34%
Introduction
抗体设计在治疗用途和生物研究中具有重要价值。然而,现有基于深度学习的方法存在以下主要问题:
- 不完整的上下文信息:现有方法在生成互补决定区(CDRs)时,仅考虑相同抗体链的骨架上下文,而没有包括目标抗原和其他抗体链的信息。这种不足可能导致无法捕捉抗体设计中重要的性质,如结合亲和力。
- 难以捕捉输入结构的完整3D几何特性:现有方法通常将3D坐标预处理为某种不变特征,这种方式丢失了特征空间中方向相关的信息,导致在描述抗体/抗原中不同残基的空间接近性方面的效率降低。此外,许多生成模型以自回归方式逐一预测CDR序列,这种方法效率低下,并在推理阶段累积误差。
为了解决这些问题,本文将抗体设计表述为 E(3)等变图翻译 问题,并提出了以下主要贡献:
- 新任务:将生成任务扩展为条件生成任务,输入不仅包含CDRs的重链上下文,还包括抗原和轻链的信息。
- 新模型:提出了一种端到端的 多通道等变注意力网络(MEAN),能够同时生成CDRs的一维序列和三维结构。MEAN直接操作在三维坐标空间中,具有E(3)等变特性,能够完整地保留残基的几何信息。通过在内部上下文编码器和外部注意力编码器之间交替,MEAN利用3D信息传递和等变注意力机制捕捉输入复杂体内不同组件之间的长程和复杂空间交互。
- 高效预测:提出了一种逐步生成CDRs的 全幅解码策略(progressive full-shot decoding),每轮生成同时更新序列和结构,避免了传统自回归模型中的误差累积,并在推理阶段提升了效率。
实验结果表明,MEAN模型在以下任务上显著优于现有的最先进方法:
- 序列和结构建模;
- 抗原结合CDR设计;
- 结合亲和力优化。
具体而言,在抗原结合CDR设计中,MEAN比基线方法的相对提升约为23%;在亲和力优化中,相对提升约为34%。
Method
Preliminaries, Notations and Task Formulation
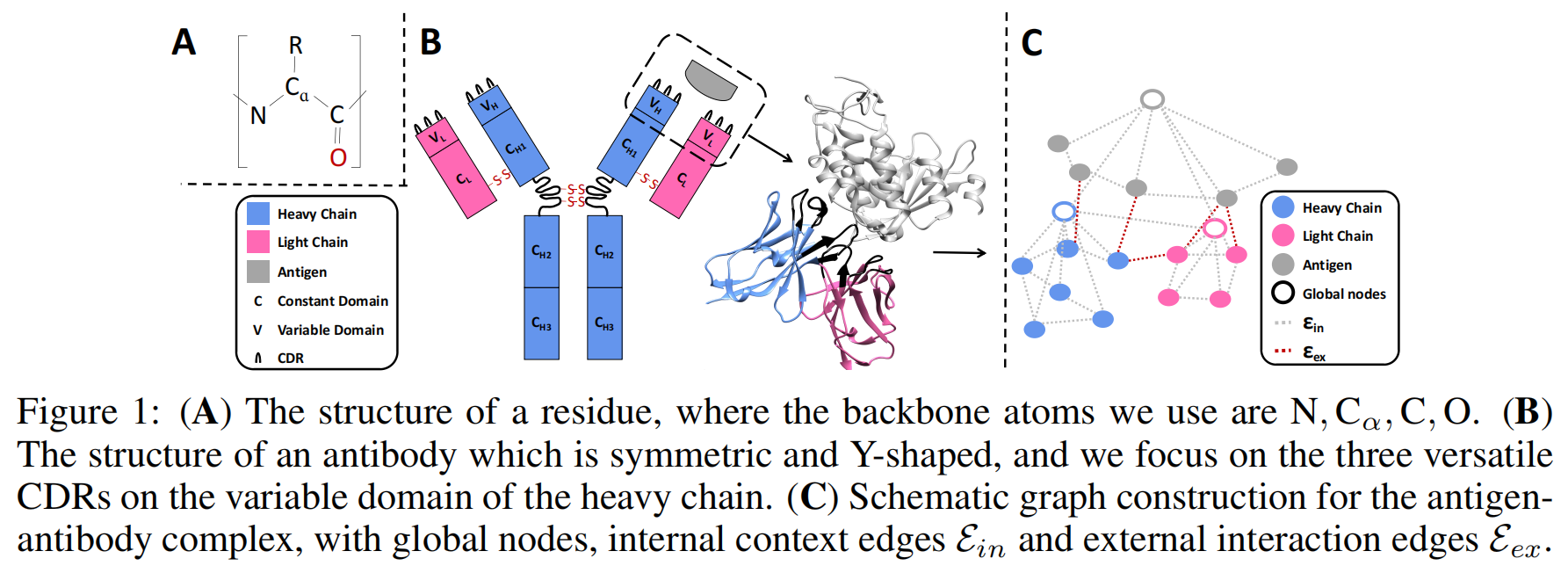
抗体是“Y”形的蛋白质,由两对对称的链组成,每一对链包括一条重链和一条轻链。在每条链中,有一些常数结构域和一个可变结构域(VH / VL),其中有三个互补决定区(CDRs)。抗体的抗原结合位点位于可变结构域中,特别是在CDR-H3中。其他部分称为框架区,通常在结构上较为保守。
任务定义:
本文的抗体设计任务是生成能与给定抗原结合的CDRs。传统的抗体设计方法专注于在重链的框架区中寻找合适的CDRs。与这些方法不同,本文不仅考虑重链的上下文,还结合抗原和轻链的信息,以便更好地控制生成抗体的结合特性。
图表示法:
我们将每个抗体-抗原复合物表示为一个图,图中包含三个空间聚集的组件,分别是重链(VH)、轻链(VL)和抗原(VA)。图G = (V, E)中的节点V包含重链、轻链和抗原的残基,边E分为内部边(Ein)和外部边(Eex)。其中,Ein表示同一组件内的节点连接,而Eex表示跨组件的节点连接。
每个节点i的特征由两个部分组成:
- hᵢ:一个可训练的特征向量,表示氨基酸类型。
- Zᵢ:一个包含多个背骨原子坐标的矩阵。
对于生成的CDRs(即重链中的CDRs部分),我们将其初始化为掩码向量,并根据CDR前后的残基位置来分布坐标。
边的构建:
- 内部边(Ein):
连接同一组件内的节点。如果两个残基之间的空间距离小于设定的阈值c₁,则建立边。相邻的残基总是建立边,以便捕获局部序列关系。 - 外部边(Eex):
连接不同组件之间的节点,如果它们之间的距离小于设定的阈值c₂(c₂ > c₁)。这些边代表抗体链和抗原之间的相互作用,主要通过分子间的力传递。
全局节点(Global Nodes):
为了让生成的CDRs充分了解其所处链的整体结构,我们引入了全局节点,并将这些节点与组件内的其他节点相连。每个全局节点的坐标由相应链的可变结构域的所有坐标的均值来确定。
MEAN: Muti-Channel Equivariant Attention Network
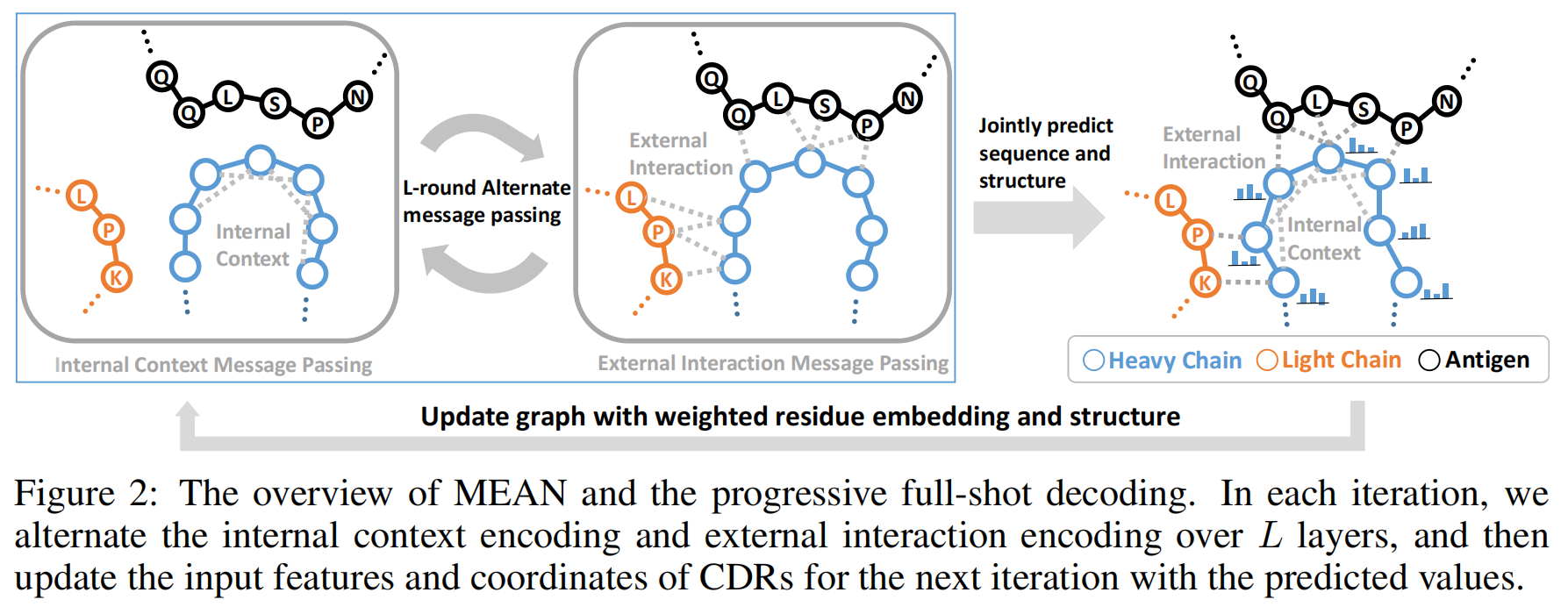
核心思想:
MEAN模型通过在每一层交替使用两个模块来捕获抗体-抗原复合物中不同组件之间的几何和拓扑信息:
- 内部上下文编码器(Internal Context Encoder):
用于处理同一组件内的节点交互,捕获组件内部的结构信息。 - 外部注意力编码器(External Attention Encoder):
用于处理跨组件之间的节点交互,捕获抗体和抗原之间的相互作用。
每一层的工作流程包括:
- 信息传递:
对于每个节点,MEAN计算与邻居节点之间的E(3)-等变消息,并将该消息用于更新节点的特征向量和坐标。 - 节点更新:
每一层后,节点的特征向量和坐标被更新,进入下一轮处理。
内部上下文编码器:
这个编码器主要通过E(3)-等变图神经网络(EGNN)来更新节点信息。节点的特征更新包括:
- 计算与邻居节点之间的消息(mᵢⱼ)。
- 通过多层感知机(MLP)更新节点的特征(hᵢ)和坐标(Zᵢ)。
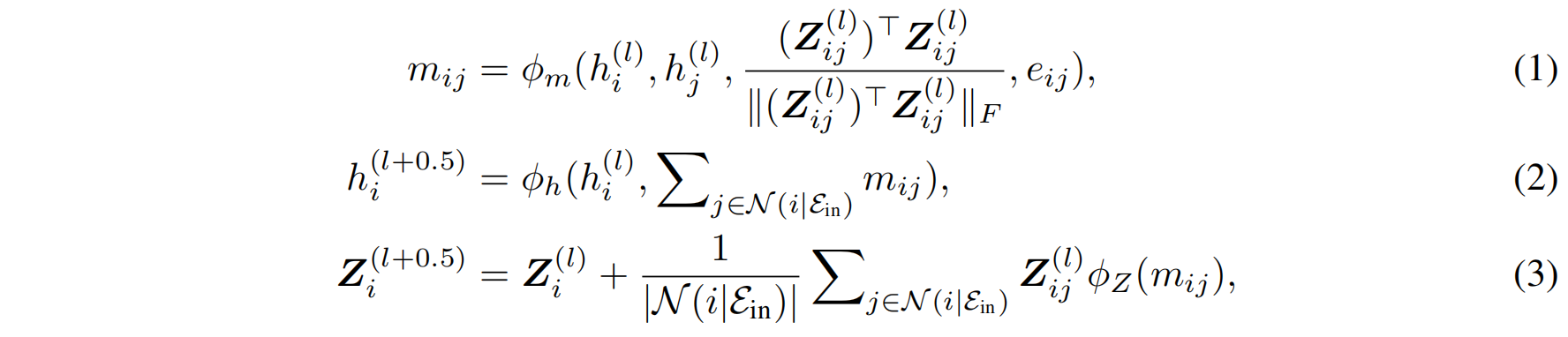
(l + 0.5)表示在该层中将由外部注意编码器进一步更新的特征和坐标
外部注意力编码器:
这个模块使用图注意力机制(Graph Attention Mechanism)来描述不同组件之间残基的相关性。与传统的注意力机制不同,我们提出了一种新的E(3)-等变图注意力机制,保证了在3D空间中信息的准确传递。具体步骤包括:
- 计算查询(qᵢ)、键(kᵢⱼ)和值(vᵢⱼ)向量。
- 计算注意力权重(αᵢⱼ),并更新节点的特征和坐标。
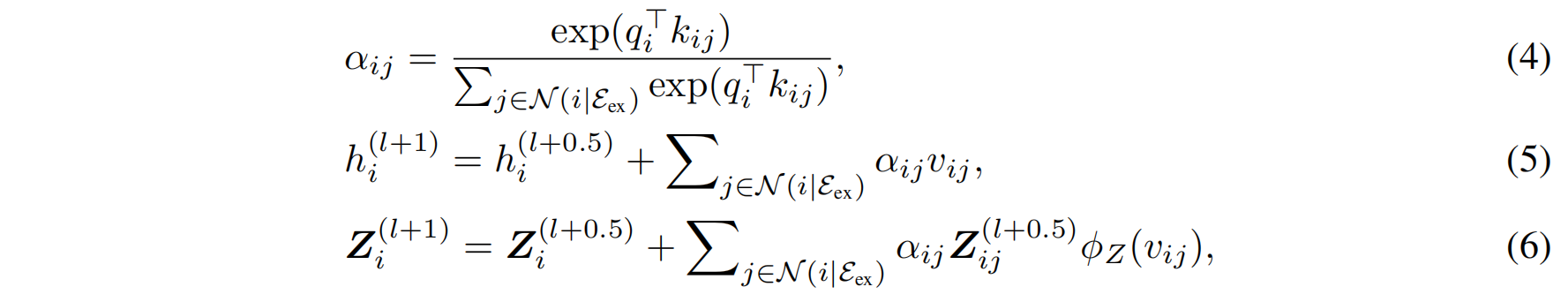
输出模块:
在所有信息传递层之后,MEAN通过输出模块计算最终的节点特征和坐标,并预测每个残基的氨基酸类型。使用SoftMax函数计算氨基酸类型的概率分布。
Progressive Full-Shot Decoding
传统方法的不足:
传统的生成方法(如RefineGNN)使用自回归方式逐一生成CDR序列,这会导致生成的错误随着时间的推移而累积。为了避免这种问题,MEAN提出了逐步全幅解码策略。
全幅解码过程:
Progressive Full-Shot Decoding 策略的核心思想是通过多轮(而非一次性)生成来提高生成质量和效率。在每一轮中,模型不再逐个生成氨基酸或三维坐标,而是同时更新所有节点的信息,包括CDR的氨基酸序列和三维结构。
具体步骤:
-
初始输入和第一轮生成:
- 输入为抗体-抗原复合物的图,包括重链、轻链和抗原的特征。CDRs部分的初始特征被设定为掩码向量,并根据附近残基的坐标来初始化。
- 在第一轮生成时,模型通过内部上下文编码器和外部注意力编码器对所有节点进行信息传递,更新每个残基的特征(氨基酸类型)和坐标(三维位置)。
-
逐步更新:
- 在每一轮的解码中,模型通过更新所有CDR残基的特征和坐标,使得整个结构逐渐完善。具体来说,模型首先根据当前轮次生成的结果(包括氨基酸分布和三维坐标)来更新节点的嵌入表示(hᵢ)和坐标(Zᵢ)。
- 对所有节点进行全幅更新时,模型利用加权嵌入策略而非选择最大概率氨基酸,这样可以减少推理过程中的误差累积。加权策略通过将每个氨基酸类别的概率分布与其嵌入表示进行加权求和,避免了选择过程中可能带来的错误。
-
轮次循环:
- 逐步生成的过程中,每一轮的更新结果都会影响下一轮的输入。这种逐步的反馈机制使得生成结果在每一轮中都能逐渐变得更加精确和稳定。
-
最终输出:
- 在最后一轮,模型输出最终的CDR序列和三维结构。经过多轮更新,结果能够更好地捕捉到结构和序列之间的相互作用,使得生成的抗体更加准确。
损失函数:
-
序列损失(Lₛₑq): 使用交叉熵计算生成的氨基酸序列与真实序列之间的差异。
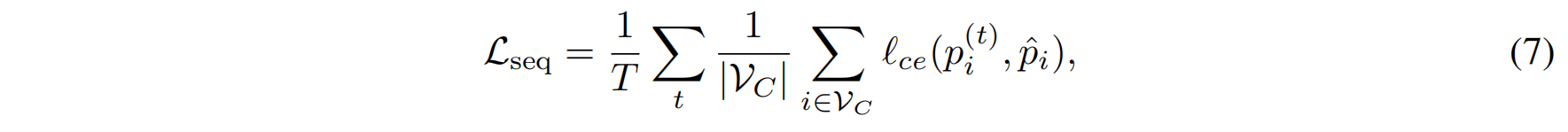
-
结构损失(Lₛₜʳᵤcₜ): 使用Huber损失(Huber Loss)计算生成的坐标与真实坐标之间的差异。该方法相较于均方误差(MSE)具有更好的鲁棒性,能够应对噪声数据。
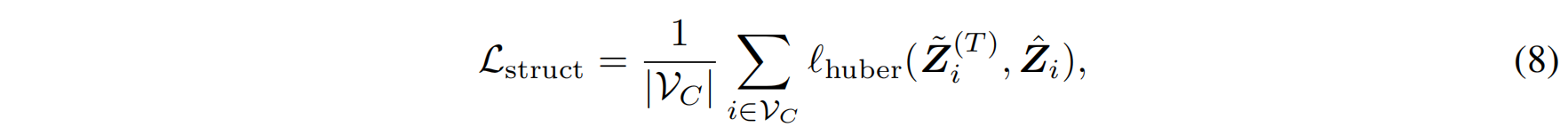
最终的损失函数是序列损失和λ结构损失的加权和
Experiments
Sequence and Structure Modeling
任务描述:
实验的目标是评估MEAN模型在生成抗体的1D序列(氨基酸序列)和3D结构(空间坐标)方面的能力。作者使用了 SAbDab(Structural Antibody Database) 数据集,该数据集包含3,127个抗体-抗原复合物。每个抗体包含多个互补决定区(CDRs),实验中重点评估了这些CDRs的1D序列和3D结构的恢复能力。
评估指标:
- Amino Acid Recovery (AAR):氨基酸恢复率,即预测的1D序列与真实序列的重叠度。
- Root Mean Square Deviation (RMSD):3D结构预测的平均平方误差,用于衡量预测结构与真实结构之间的距离。
实验设置:
- 数据预处理: 数据集中的每个抗体被按CDR类型(CDR-H1、CDR-H2、CDR-H3)进行聚类。每个CDR的聚类与MMseqs2进行序列相似性搜索,确保每个测试集中的CDR序列不与训练集中的序列过于相似。
- 数据分割: 按照8:1:1的比例将数据划分为训练集、验证集和测试集。
实验结果:
tab1
实验结果表明,MEAN显著优于其他基线方法(如LSTM、RefineGNN等),在1D序列和3D结构建模任务中取得了更高的氨基酸恢复率(AAR)和更低的RMSD
Antigen-Binding CDR-H3 Design
任务描述:
该实验评估了MEAN在设计结合特定抗原的CDR-H3时的表现。设计任务的目标是生成能够高效绑定给定抗原的CDR-H3序列,并与真实结构匹配。
评估指标:
- Amino Acid Recovery (AAR):氨基酸恢复率,评估生成的序列与真实序列的匹配程度。
- TM-score:用于衡量蛋白质结构之间的相似性,范围从0到1,值越大表示结构越相似。
- RMSD:3D结构之间的均方根偏差,评估生成结构与真实结构的差异。
实验设置:
实验中,作者从 RAbD(Antigen-Antibody Database) 中选取了60个抗体-抗原复合物进行设计,并将其作为测试集。训练集仍然使用 SAbDab 数据集,训练的集群/抗体数量为1443/32638,160/339进行验证。通过生成多个候选CDR-H3序列,作者对比了生成序列与目标抗原结合的能力。
实验结果:
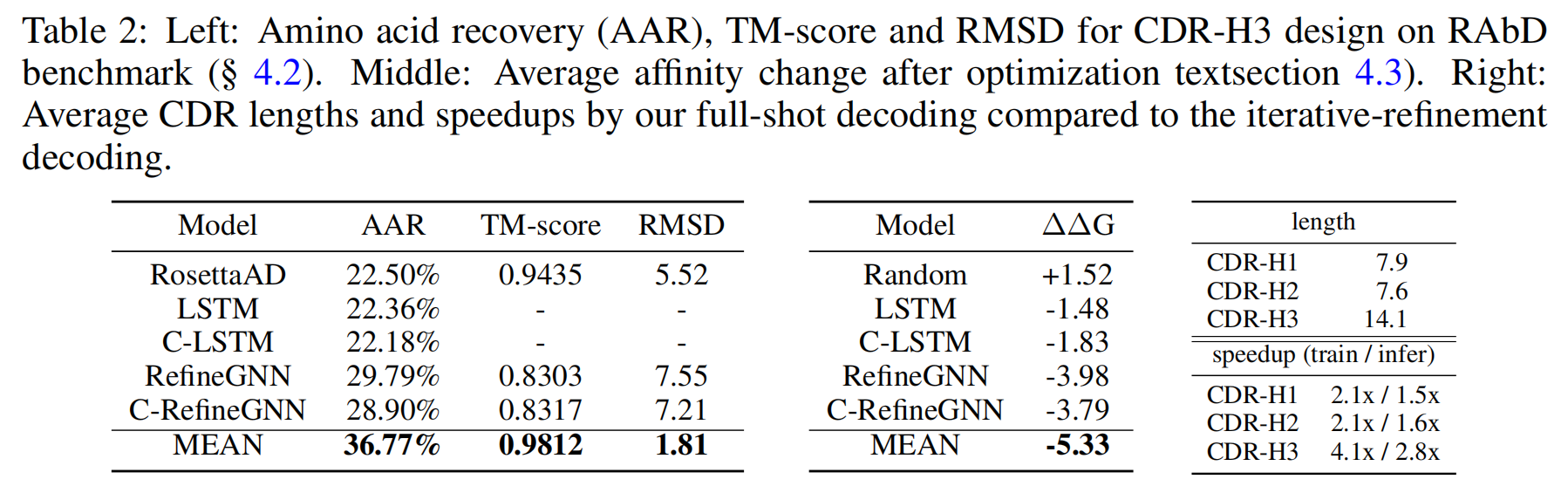
实验表明,MEAN在抗原结合设计任务中优于所有其他方法,包括经典的RosettaAD方法。MEAN在AAR和TM-score上都表现优异,尤其在TM-score上,MEAN接近于0.99,表明生成的结构与真实结构几乎完全重合
Affinity Optimization
任务描述:
该实验评估了MEAN在优化抗体亲和力方面的能力,目标是通过联合优化CDR-H3的序列和结构来提高抗体与抗原的结合亲和力。
评估指标:
- ∆∆G(变化的结合能):衡量亲和力优化的效果,∆∆G值越低表示亲和力越强。
实验设置:
作者使用 SKEMPI V2.0(Structural Kinetic and Energetic Mutant Protein Interactions)数据库来进行亲和力优化。实验中,作者通过 Iterative Target Augmentation (ITA) 算法进行优化,该算法通过生成多个候选结构并选择最佳的抗原-抗体结合体来实现亲和力优化。
实验结果:
实验表明,表2右侧,MEAN显著改善了抗体的亲和力,相较于其他方法,MEAN能产生具有更低亲和力变化(更强结合力)的抗体。
CDR-H3 Design with Docked Template
任务描述:
该实验探讨了如何利用已知的抗体-抗原结合模板来设计新的CDR-H3。目标是通过将已知的抗体框架与抗原结合模板结合,生成具有高亲和力的新CDR-H3序列。
实验设置:
作者使用 HDOCK 工具对抗原进行对接,生成抗体-抗原复合物的模板,然后利用MEAN模型生成新的抗原结合CDR-H3序列,并评估其亲和力。
实验结果:
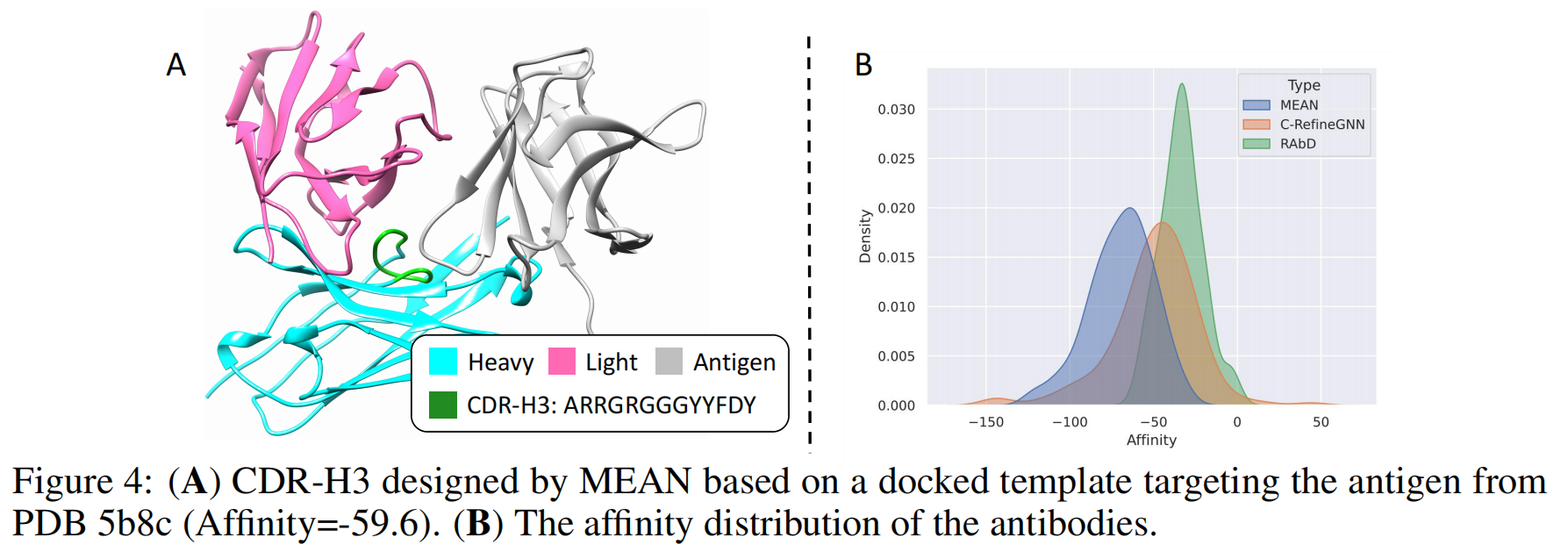
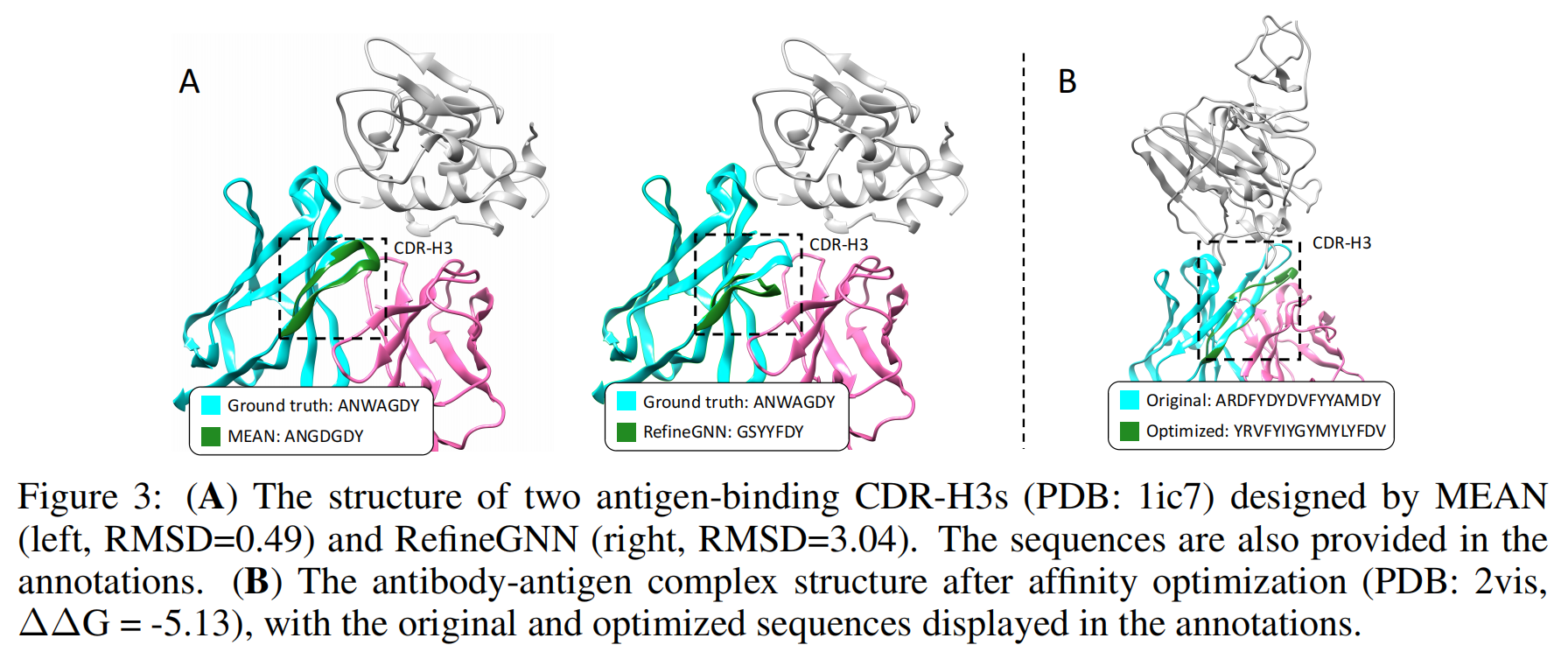
通过基于对接模板的生成,MEAN能够生成比C-RefineGNN和原始抗体更具亲和力的抗体。具体结果如下:
- 图示:作者展示了由MEAN设计的CDR-H3序列与原始结构在亲和力上的明显改进。优化后的抗体表现出显著提高的亲和力。
Analysis
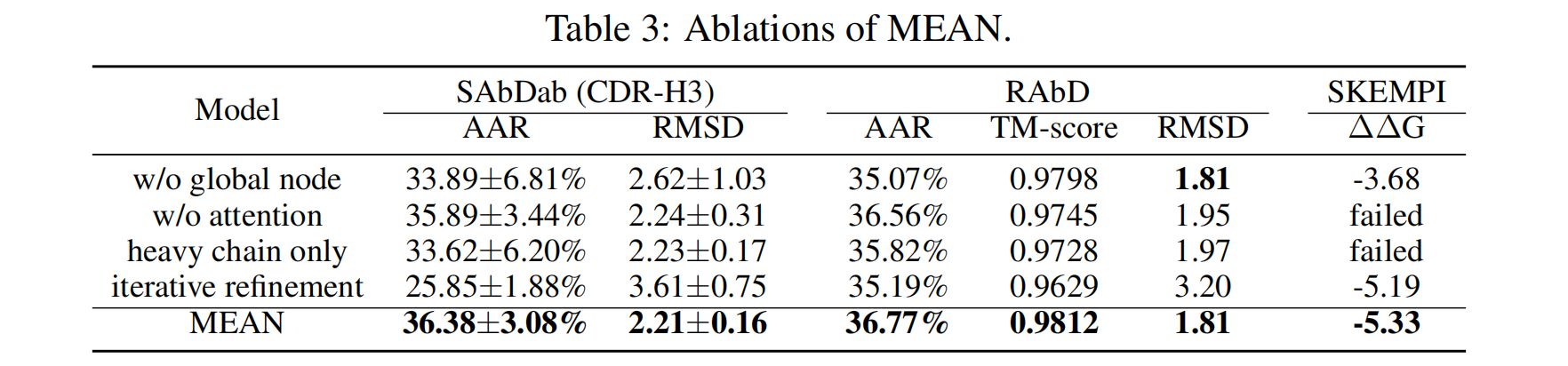
为了验证MEAN模型中的各个组成部分是否必要,作者进行了消融实验,结果显示:
- 移除全局节点或注意力机制都会导致性能下降。
- 只使用重链会显著削弱性能,表明考虑抗原和轻链的上下文信息至关重要。
- 与逐步全幅解码相比,使用迭代优化方法的模型性能较差,证明逐步全幅解码策略在计算效率和生成质量方面的优势。

