论文地址:Multi-Scale Representation Learning for Protein Fitness Prediction
S3F:序列+结构+点云突变预测
Abstract
该论文研究了如何通过多尺度表示学习改进蛋白质适应度(fitness)预测。设计新功能蛋白的关键在于准确建模其适应度景观(fitness landscape),但由于实验数据稀缺,现有方法主要依赖于在大规模、无标签的蛋白质序列或结构数据上训练的自监督模型。尽管早期的蛋白质表示学习研究仅关注序列或结构特征,近期的混合架构试图结合这两种模态,以利用各自优势。然而,现有的序列-结构模型仅在性能上取得了有限的提升,表明如何高效融合这些模态仍然是一个挑战。此外,某些蛋白质的功能高度依赖于其表面拓扑的精细特征,这些特征在此前的模型中被忽略。为了解决上述问题,该研究提出了S3F(Sequence-Structure-Surface Fitness)模型,一种新型的多模态表示学习框架,能够在多个尺度上整合蛋白质特征。该方法结合了蛋白质语言模型生成的序列表示、Geometric Vector Perceptron(GVP) 网络编码的蛋白质主链结构信息,以及表面拓扑的精细描述。实验表明,该方法在ProteinGym 基准(包含 217 个深度突变扫描实验)上达到了最先进的适应度预测性能,并提供了对蛋白质功能决定因素的深入理解。
1 Introduction
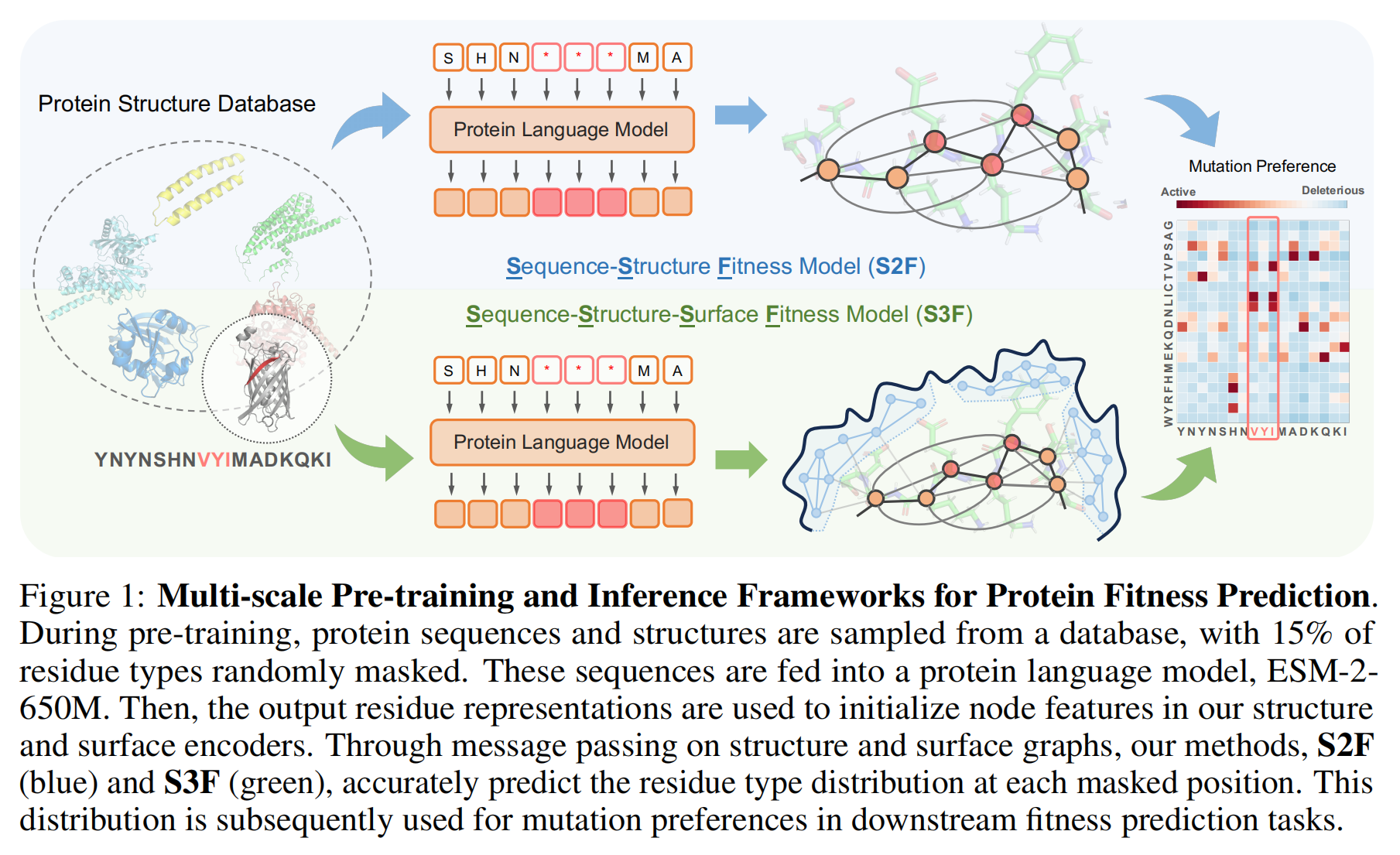
蛋白质在自然界中执行着多种功能,例如催化化学反应、维持细胞结构、运输分子和传递信号。这些功能由蛋白质的氨基酸序列和三维结构决定。通过优化这些序列和结构,人们可以在可持续发展、新材料和医疗健康等领域解决关键挑战。而这一优化过程的起点是学习蛋白质序列或结构与其功能之间的关系,即适应度景观(fitness landscape)。适应度景观是一个多变量函数,描述了突变对蛋白质适应度的影响——越准确地建模这些景观,越有可能设计出具有所需特性的蛋白质。
然而,适应度建模面临的主要挑战是实验收集的功能性标签极为稀缺,而蛋白质的可能序列空间却极为庞大。因此,自监督学习(self-supervised learning)*成为了一种重要的策略,利用海量的无标注蛋白质数据来学习突变效应的预测方法。早期的方法通常通过*多序列比对(MSA) 来学习特定蛋白质家族的序列分布,而后续研究则尝试学习跨家族的通用功能模式,催生了蛋白质语言模型(protein language models)** 或家族无关(family-agnostic)模型。近期的混合方法尝试结合这两类方法的优势,达到了最先进的适应度预测性能。
尽管基于序列的方法可以部分恢复蛋白质的结构信息,但许多关键的蛋白质功能和任务依赖于更精细的结构和表面特征。为此,近年来的研究开始探索蛋白质结构表示学习,例如逆折叠(inverse folding) 方法可以在已知蛋白主链结构的情况下学习其序列分布,并已在稳定性预测等任务上展现出优越性能。此外,近期的研究还尝试结合序列和结构方法,例如AlphaMissense 通过引入结构预测损失,将结构信息蒸馏到混合模型中,展现了结构特征的价值。然而,这些混合方法仅取得了有限的提升,或未公开其模型权重。此外,现有方法难以有效建模蛋白质表面,而蛋白质表面对其相互作用至关重要,并能提供更丰富的结构信息。
本研究提出了一种多尺度蛋白质表示学习框架,用于零样本(zero-shot)蛋白质适应度预测。首先,提出S2F(Sequence-Structure Fitness)模型,结合蛋白质语言模型和结构编码器,其中结构编码器采用Geometric Vector Perceptron(GVP),在空间邻域间进行消息传递。进一步地,研究提出S3F(Sequence-Structure-Surface Fitness)模型,在 S2F 的基础上引入蛋白质表面编码器,通过点云(point cloud)表示蛋白质表面,并进行表面点之间的消息传递。这些多尺度蛋白质编码器基于CATH 数据集(蛋白结构分类数据库)上的残基类型预测任务进行预训练,使其能够直接进行突变效应的零样本预测。
本研究的贡献包括:
- 提出了一种通用且模块化的框架,用于学习蛋白质的多尺度表示;
- 提出了两种具体模型——S2F(结合序列和结构)和 S3F(结合序列、结构和表面),用于提升蛋白质适应度预测性能;
- 在 ProteinGym 基准测试上进行严格评估(包含 217 个深度突变扫描实验),并在适应度预测任务上达到了当前最优性能,同时提高了预训练效率;
- 针对不同类型的实验进行深入分析,展现了多尺度学习的稳定提升,并证明结合结构和表面特征可以纠正基于序列的方法的偏差,提升结构相关功能的准确性,并增强对上位效应(epistasis) 的建模能力。
3 Method
本研究提出了一种多尺度蛋白质表示学习框架,用于零样本(zero-shot)蛋白质适应度预测。该方法结合了序列、结构和表面三种蛋白质特征,并提出了两个具体的实例模型:S2F(Sequence-Structure Fitness Model) 和 S3F(Sequence-Structure-Surface Fitness Model),前者结合了蛋白质序列与结构信息,后者进一步整合了蛋白质表面特征。
3.1 Preliminary
蛋白质表示:
一个蛋白质由 $n_r$ 个残基(氨基酸)和 $n_a$ 个原子组成,可以表示为一个序列-结构二元组 $(S, X)$:
- 序列(Sequence) $S = [s_1, s_2, …, s_{n_r}]$,其中 $s_i$ 代表第 $i$ 个残基的类型(20 种可能的氨基酸)。
- 结构(Structure) $X = [x_1, x_2, …, x_{n_a}] \in \mathbb{R}^{n_a \times 3}$,其中 $x_i$ 代表第 $i$ 个原子的三维坐标。为简化建模,仅使用 主链的 α-碳原子,忽略侧链变化。
蛋白质适应度景观(Fitness Landscape):
蛋白质的功能取决于其序列和折叠结构。序列的突变会改变适应度,该关系可以通过深度突变扫描(DMS, Deep Mutational Scanning) 实验测量。
问题定义:
研究目标是在无监督的设置下预测突变对蛋白质适应度的影响。具体而言:
- 给定一个野生型蛋白 $(S_{wt}, X_{wt})$;
- 通过突变产生突变体 $(S_{mt}, X_{mt})$,其中在突变位点 $T$ 处的残基改变 $s_{mt}^t \neq s_{wt}^t$;
- 假设主链结构保持不变(即 $X_{mt} = X_{wt}$);
- 目标是设计一个无监督模型,为突变体分配一个适应度得分,衡量其相对于野生型的适应度变化。
3.2 Protein Language Models for Mutational Effect Prediction
蛋白质语言模型(如 ESM)采用掩码语言建模(masked language modeling, MLM) 进行训练,学习残基的共现模式。给定一个序列 SSS,突变效应可以使用对数几率比(log odds ratio) 计算:
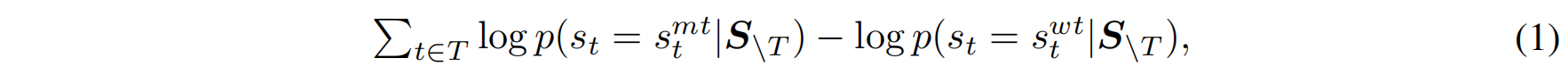
其中,$S_{\backslash T}$ 表示在突变位点 $T$ 处进行掩码的输入序列,假设多个突变之间相互独立。在零样本(zero-shot)情况下,模型仅通过前向传播进行推理,而无需额外训练。
3.3 Sequence-Structure Model for Fitness Prediction
问题:
蛋白质语言模型忽略了蛋白质的三维结构信息,而结构信息对于蛋白质功能至关重要。因此,S2F 通过结合蛋白质序列和结构来提升适应度预测能力。
模型假设:
- 主链结构在突变后保持不变(不建模突变如何影响结构)。
- 忽略侧链信息,因为侧链直接暴露了残基类型,可能导致信息泄漏。
几何消息传递(Geometric Message Passing):
-
构造半径图(radius graph): 以 α-碳 为节点,若两个残基的欧几里得距离小于 10Å,则连边。
-
使用 GVP(Geometric Vector Perceptron)
进行消息传递:
-
GVP 结合标量和向量特征,保证旋转和平移不变性(SE(3)-等变)。
-
初始节点特征使用蛋白质语言模型(ESM) 提供的序列嵌入。
-
通过 5 层 GVP 进行消息传递,更新节点表示:
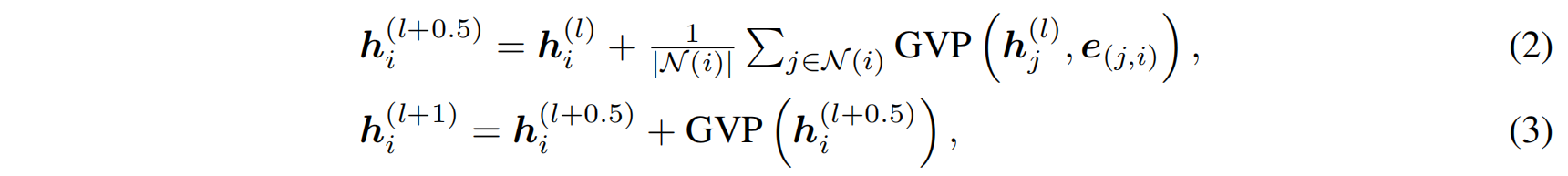
-
3.4 Sequence-Structure-Surface Model for Fitness Prediction (S3F)
问题:
除了序列和结构,蛋白质的表面特征对功能也至关重要。例如,亲水和疏水残基在折叠后会分别暴露在溶剂中或埋藏在蛋白质核心中。因此,S3F 进一步加入蛋白质表面信息。
表面处理:
- 使用 dMaSIF 方法基于主链结构生成蛋白质表面,并以点云(point cloud) 形式表示,每个蛋白表面包含 6K-20K 个点。
- 每个点 iii 具有几何特征 fif_ifi,包括高斯曲率和热核签名(Heat Kernel Signature),用于捕获表面拓扑。
表面特征初始化:
-
通过最近邻搜索,将表面点的特征与其最近的 3 个残基(从 S2F 中获得嵌入)结合,初始化表面点表示:
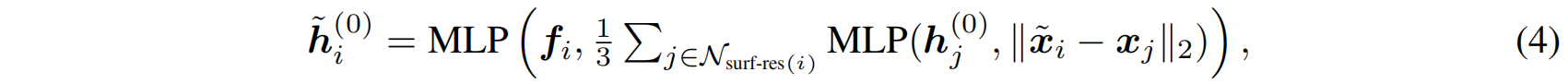
表面消息传递:
-
构造表面 KNN 图(k = 16),使用 GVP 进行消息传递,与 S2F 的结构图类似:
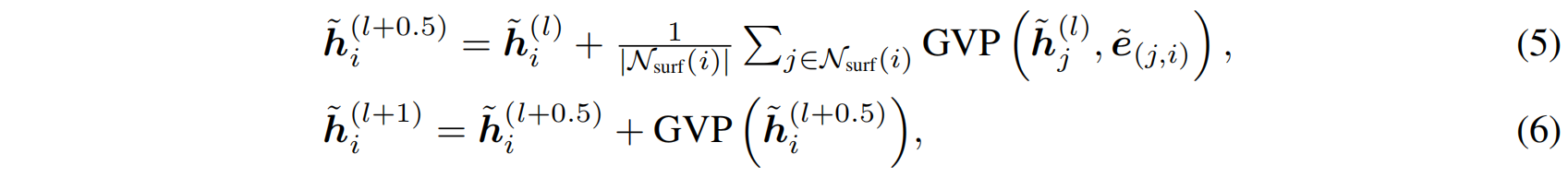
残基表示融合(Residue Representation Aggregation):
-
结构图(S2F)和表面图(S3F)的最终嵌入进行融合:
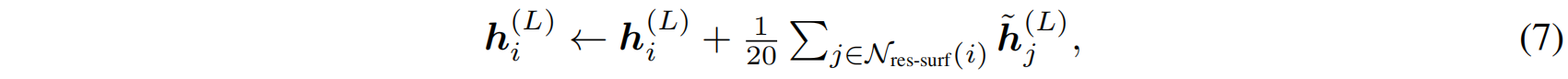
3.5 Pre-Training and Inference
预训练任务:残基类型预测(masked residue type prediction),随机掩盖 15% 的残基,并用交叉熵损失 进行训练。
预训练数据:CATH 数据集(30,948 个实验结构),模型仅训练 GVP 层,而ESM-2-650M 权重冻结。
推理时,使用 AlphaFold2 预测野生型结构,并根据结构质量(pLDDT 分数)决定是否使用 S3F 预测适应度分数。
4 Experiment
4.1 Setup(实验设置)
评估数据集
实验使用 ProteinGym 基准测试(MIT License),该数据集包含 深度突变扫描(DMS, Deep Mutational Scanning)实验,涵盖:
- 功能类别(如稳定性、配体结合、病毒复制、耐药性等)。
- 217 个替换(substitution)突变实验,包括单点突变和多点突变。
评估指标
适应度预测面临高度非线性的问题,因此实验主要使用 Spearman’s 相关系数(Spearman’s rank correlation) 评估预测结果与实验测量值的一致性。此外,研究还报告:AUC(ROC 曲线下的面积),MCC(Matthews 相关系数),NDCG(Normalized Discounted Cumulative Gain),Top 10% Recall(识别最高适应度突变的能力)
对比基线
- 无 MSA 方法(不使用多序列比对信息):
- 蛋白质语言模型:ProGen2 XL, CARP, ESM-2-650M
- 逆折叠模型:ProteinMPNN, MIF, ESM-IF
- 序列-结构混合模型:MIF-ST, ProtSSN, SaProt
- 使用 MSA 的方法:
- 家族特定(family-specific)模型:DeepSequence, EVE, GEMME
- 混合(family-agnostic + MSA)模型:MSA Transformer, Tranception L, TranceptEVE
此外,研究进一步结合 EVE 预测结果,构造两种增强版本:
- S2F-MSA(S2F + EVE 预测融合)
- S3F-MSA(S3F + EVE 预测融合)
4.2 Benchmark Result
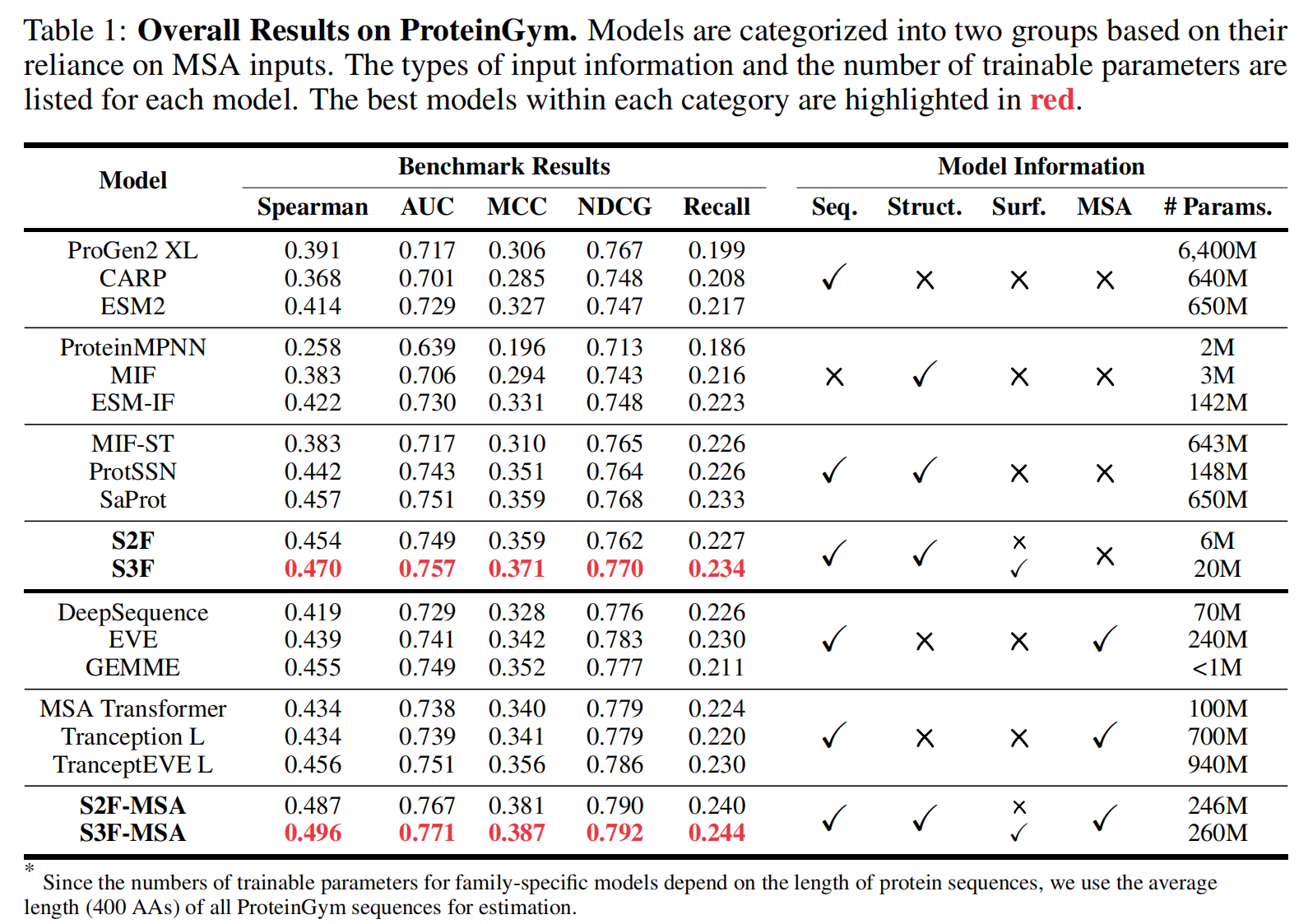
实验结果总结在 Table 1,其中:
- S2F 取得了与现有最优方法相当的性能,超过了大多数蛋白质语言模型和逆折叠模型,仅略低于 SaProt。
- S3F 通过加入蛋白质表面信息,成为无 MSA 方法中的最佳模型,其 Spearman’s 相关系数(0.470)超过了 TranceptEVE(0.456),后者是使用 MSA 的最先进方法。
- 参数量较小但性能卓越:S3F 仅有 20M 训练参数,相较于 SaProt(650M),其预训练时间大幅缩短。
- S3F-MSA 进一步结合 MSA 信息,将 Spearman’s 提升 8.5%(0.496),超越了当前最先进的 SaProt 方法。
4.3 Breakdown Analysis for Multi-Scale Learning
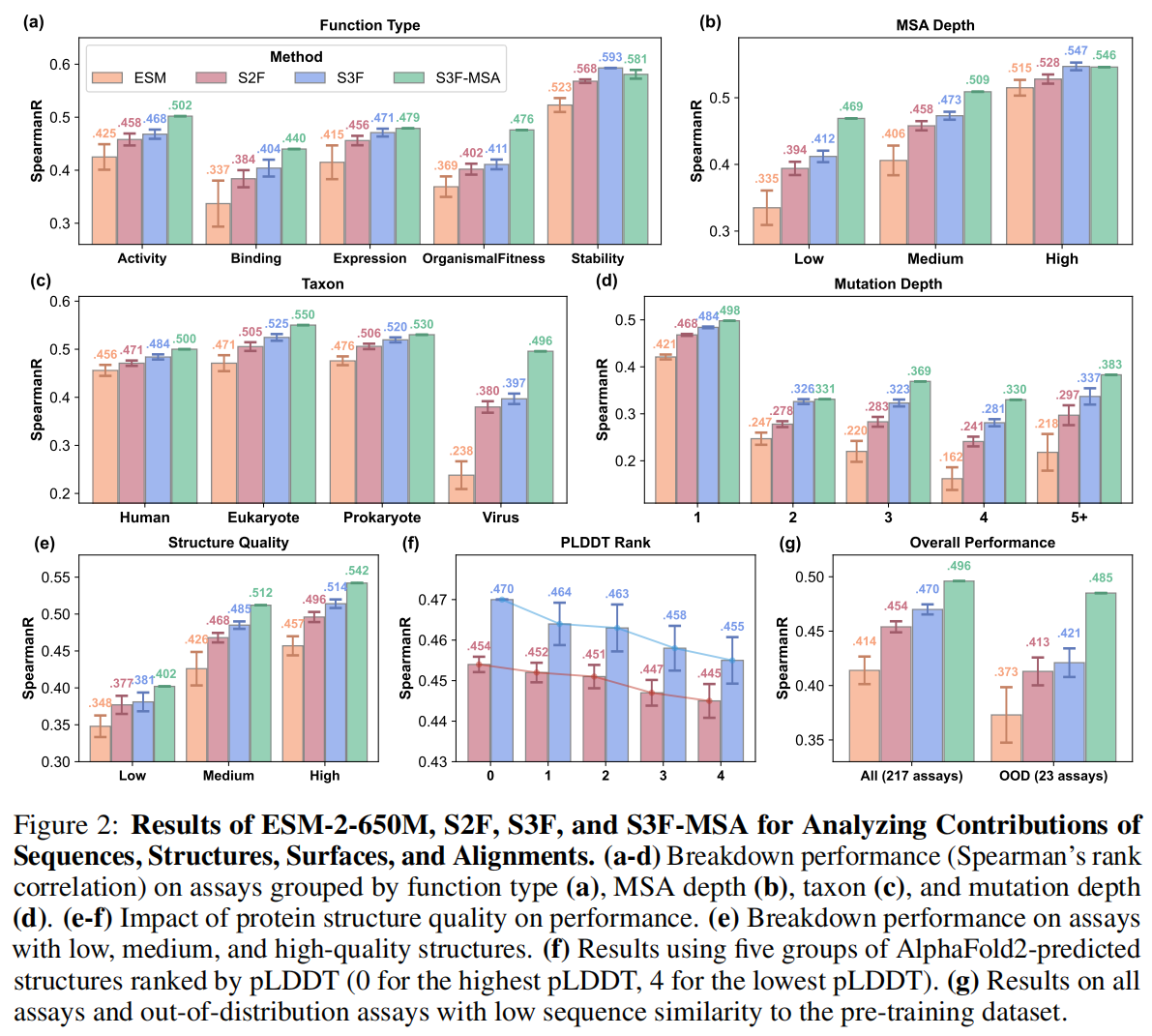
为进一步理解多尺度表示学习的贡献,研究对不同类型的实验进行细分分析,Figure 2 (a-d) 详细展示了 ESM-2-650M、S2F、S3F 和 S3F-MSA 在不同实验条件下的 Spearman’s 相关系数。
- 功能类别(Figure 2a):
- 结构和表面特征对结合(binding)和稳定性(stability)任务的提升尤为显著。
- 这与直觉一致,因为这些功能通常依赖于蛋白质的几何结构,结构信息有助于识别破坏结构的突变。
- MSA 深度(Figure 2b):
- 在低 MSA 深度的蛋白质上,ESM 的表现较差,表明蛋白质语言模型的预训练数据集中缺乏此类蛋白。
- S2F 和 S3F 通过结构特征缓解了这一问题,在低 MSA 深度的蛋白质上提供更强的预测能力。
- 物种类别(Figure 2c):
- 在病毒蛋白上,ESM-2-650M 的表现较差,而 S3F 明显提高了 Spearman’s 相关系数,表明结构和表面信息有助于解决序列模型的偏差。
- 突变深度(Figure 2d):
- 单点突变(single mutation)比多点突变(multiple mutations)表现更优,这与模型假设(突变效应的加性)一致。
- 随着突变深度增加,S3F 的改进更明显,表明结构和表面信息有助于建模上位效应(epistasis)。
4.4 Impact of Structure Quality
由于许多蛋白质没有实验确定的结构,本研究使用 AlphaFold2 预测蛋白质结构,并分析其质量对模型表现的影响。
- 按结构质量分类(Figure 2e):
- 研究将 217 个实验按照 pLDDT(AlphaFold 结构质量评分)分为**高(>90)、中(70-90)、低(<70)**三类。
- 结果表明:所有方法的性能都与结构质量正相关,特别是 S3F,在高质量结构上受益更显著。
- 不同质量的 AlphaFold2 结构(Figure 2f):
- 使用 5 组 pLDDT 评分不同的 AlphaFold2 预测结构进行推理,结果表明:
- 低质量结构会显著降低 S2F 和 S3F 的表现,进一步证明模型对结构质量的依赖性。
4.5 Generalization Ability to Unseen Protein Families
研究进一步考察 S3F 在未见过的蛋白质家族上的泛化能力:
- 选取 23 个蛋白质实验,其序列与预训练数据集(CATH)相似性低于 30%。
- 结果(Figure 2g)表明:
- 尽管所有方法的性能略有下降,S2F 和 S3F 仍保持稳定的改进趋势。
- 说明多尺度学习方法能够泛化到新的蛋白质家族。
4.6 Case Study: Epistatic effects in the IgG-binding domain of protein G

为了进一步分析 S3F 如何提升突变效应预测能力,研究选择了 GB1(免疫球蛋白结合域) 的突变实验进行案例研究:
-
GB1 数据集包含所有单点和双点突变的测量值,因此可以分析上位效应(epistasis)。
-
Figure 3 (a-c)
展示了 ESM、S2F、S3F 对突变对(mutation pairs)的 Spearman’s 相关性:
- S2F 和 S3F 能够更准确地预测远距离但空间接近的残基之间的相互作用,这表明结构和表面特征有助于捕捉上位效应。
- Figure 3 (d) 进一步展示了突变区域的结构,可视化了这些区域如何影响蛋白质功能。

