论文地址:Retrieval Augmented Diffusion Model for Structure-informed Antibody Design and Optimization
RADAb:DiffAb+结构检索做序列设计
Abstract
提出了一种检索增强扩散模型(Retrieval-Augmented Diffusion Model, RADM),用于结构信息驱动的抗体设计与优化。RADM 结合了检索机制和扩散模型,以提高抗体序列的生成质量,同时保证其结构稳定性和功能优化。
该方法利用一个包含已知抗体序列和结构的大规模数据库,在生成过程中检索最相关的抗体序列,并将其信息融入扩散模型的去噪过程。这种方式使模型能够借鉴已有的高质量抗体,从而提高生成序列的可行性和功能性。实验表明,RADM 在多项抗体设计任务上优于现有方法,包括序列多样性、结构保真度和功能优化能力。
1 Introduction
抗体是免疫系统中关键的 Y 形蛋白,负责识别和中和特定的抗原。这种特异性主要来源于互补决定区(CDRs),CDRs 在抗原结合亲和力方面起着至关重要的作用。因此,设计有效的 CDRs 是开发高效治疗性抗体的核心。目前,抗体的开发主要依赖于动物免疫、抗体文库筛选等劳动密集型实验方法,但这些方法成本高昂、效率较低。
近年来,深度学习和生成模型在蛋白质设计领域取得了重要进展,例如几何学习和生成建模能够直接从数据中捕捉氨基酸残基之间的高阶相互作用。然而,尽管这些方法具有强大的生成能力,当前的生成模型在抗体设计上仍面临挑战,主要问题包括:
- 结构约束的缺失:现有模型难以生成符合物理化学特性的抗体,导致设计出的抗体序列不符合天然抗体的结构规律。
- 训练数据的多样性不足:主流研究主要依赖于 SAbDab 数据库(Dunbar et al., 2014),其中包含的抗原-抗体复合体结构不足 1 万个,数据量有限,难以全面捕捉抗体与抗原之间的高阶相互作用信息,增加了模型过拟合的风险。
- 缺乏模板指导:目前大多数方法采用从头(de novo)序列生成,未能有效利用已有的抗体模板信息。这种方法需要大量数据和长时间训练,才能在实际应用中达到可接受的性能。
本文工作
受到模板驱动和片段驱动抗体设计的启发,本文提出了一种检索增强扩散抗体设计模型(Retrieval-Augmented Diffusion Antibody Design Model, RADAb)。该模型综合利用:
- 蛋白质结构数据库,检索最相关的 CDR 片段;
- 有效的结构-进化信号融合机制,防止模型过拟合;
- 半参数生成神经网络,减少训练需求,提高实际应用的可行性。
具体来说,RADM 采用以下策略:
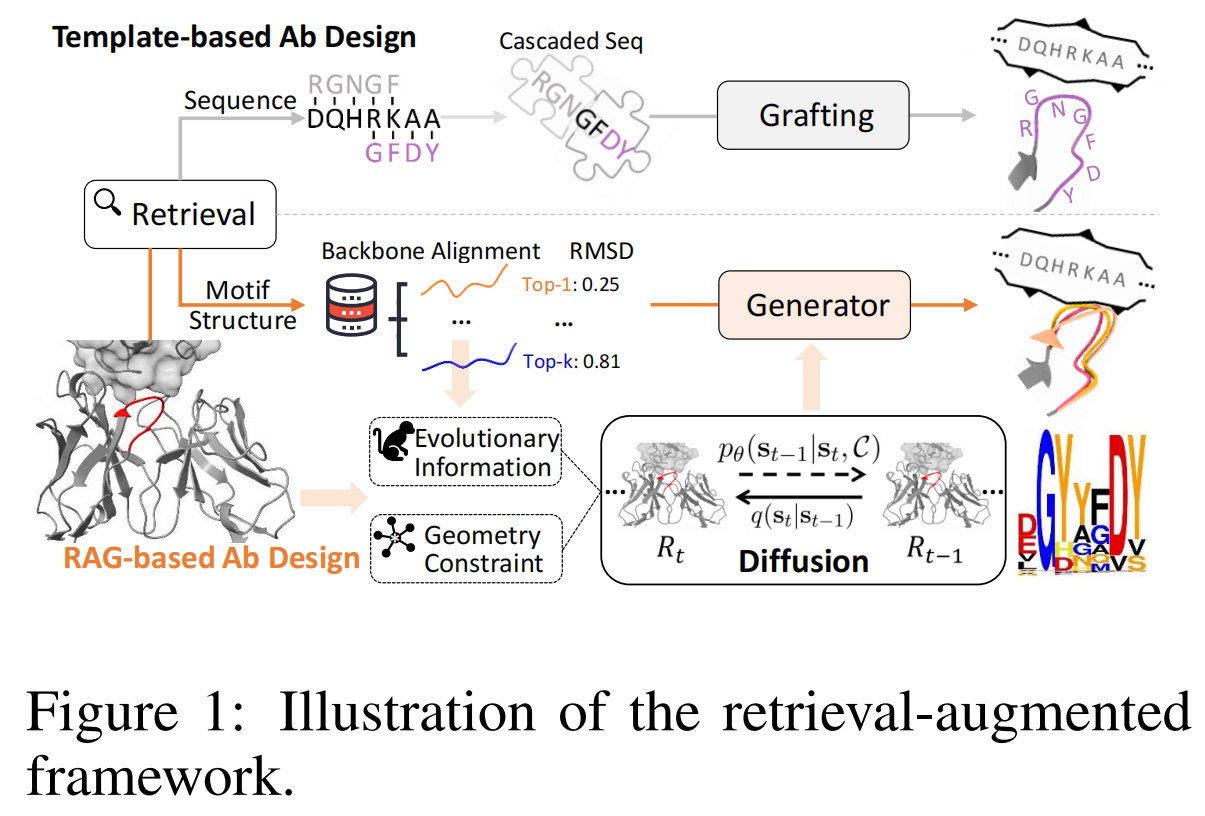
- 构建 CDR 片段数据库:从非冗余蛋白数据库(PDB)(Berman et al., 2000)中提取 CDR 片段,这些片段不仅存在于抗体结构中,也可以是 PDB 中符合 CDR 结构特征的其他蛋白质片段。
- 检索最相关的 CDR 片段:对于目标抗体序列,RADM 通过结构检索找到具有相似骨架的 CDR 片段,并假设这些片段在进化上具有相似性,可增强模型的泛化能力。
- 融合多个结构同源片段进行序列优化:不同于传统方法只利用单一 CDR 片段进行优化,RADM 采用一组结构同源的 CDR 片段,结合目标抗体的骨架进行迭代优化
主要贡献
- 首次提出基于检索增强的生成框架用于抗体设计。该框架通过一组 CDR 片段指导生成,确保序列符合目标骨架结构,并满足所有所需的生物化学性质。
- 创新性地引入检索机制,在输入抗体骨架的基础上,利用双分支去噪模块整合结构和进化信息。此外,耦合条件扩散模块(coupled conditional diffusion module)在生成过程中迭代优化,结合全局和局部信息,比传统抗体逆折叠(inverse folding)方法能捕捉到更多功能性信息。
- 实验结果表明,RADM 在多个抗体逆折叠任务上超越了现有 SOTA 方法,例如:
- 在长 CDRH3 逆折叠任务中提高了 8.08% 的 AAR(Amino Acid Recovery);
- 在功能优化任务中提升了 7 cal/mol 的绝对 ∆∆G 评分。
3 Preliminaries and Notations
3.1 Notations
抗体由两条重链(heavy chain, H)和两条轻链(light chain, L)*组成。每条链的末端都含有一个互补结合位点,该位点*特异性地与抗原(antigen)上的特定位点(表位,epitope)结合。该结合位点主要由六个互补决定区(CDRs)** 组成:
- 重链 CDRs:CDR-H1、CDR-H2、CDR-H3
- 轻链 CDRs:CDR-L1、CDR-L2、CDR-L3
CDRs 负责抗体的特异性结合,因此优化 CDR 序列是抗体设计的核心任务。
在本文中,每个氨基酸残基(residue)被表示为以下三种信息:
- 残基类型(Residue type)
- 记作 sis_isi,属于20 种天然氨基酸集合:$s_i \in { A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y }$
- 空间坐标(Coordinates)
- 记作 $x_i \in \mathbb{R}^3$,表示该残基在三维空间中的位置。
- 取向信息(Orientation)
- 记作 $O_i \in SO(3)$,表示该残基的三维旋转取向。
此外,给定一个 CDR 片段,其氨基酸序列包含 $m$ 个残基,从位置 $a$ 开始,可表示为:$R = {s_j | j \in {a+1, \dots, a+m}}$
其中,RRR 代表待优化的 CDR 序列。
整个抗体序列长度为 MMM,其抗体骨架(Antibody Framework) 由除 CDR 片段外的其余部分组成:$C_{ab} = { (s_i, x_j, O_j) | i \in {1, \dots, M} \setminus {a+1, \dots, a+m}, j \in {1, \dots, M} }$
其中:
-
抗体骨架序列(Antibody Framework Sequence):$S_{ab} = { s_i | i \in {1, …, M} \setminus {a+1, …, a+m} }$
-
抗原信息(Antigen):$C_{ag} = { (s_i, x_i, O_i) | i \in {M+1, …, N} }$
其中,抗原序列的索引范围是 $M+1$ 到 $N$。
-
检索得到的 CDR 片段(Retrieved CDR-like Fragments):$A = { A_i | i \in {1, …, k} }$
其中,kkk 代表检索得到的 CDR 片段数量。
模型目标:
- 提取抗体骨架复合物 $C_{ab}$ 中的 CDR 结构;
- 输入 CDR 结构至检索模块,获取相似的 CDR 片段 $A$;
- 结合 $C_{ag}$(抗原)、$C_{ab}$(抗体骨架)和 $A$(检索片段),预测目标 CDR 片段 $R$ 的分布。
3.2 Diffusion Model for Antibody Design
由于扩散模型(Diffusion Models, DMs)在生成任务中的出色表现和可控性,近年来,许多基于扩散的工作已经取得了显著成果(Luo et al., 2022; Villegas-Morcillo et al., 2023; Kulytė et al., 2024)。
本文关注序列生成(sequence generation),其中前向扩散过程(Forward Process)会对氨基酸序列数据进行扰动,具体如下(Hoogeboom et al., 2021):
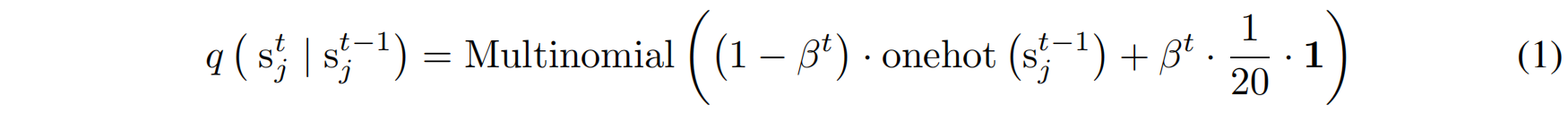
- 其中:
- $\beta^t$ 是扩散过程的噪声调度(noise schedule),当 $t$ 逐渐接近终点 $T$ 时,$\beta^t$ 会趋于 1,使得概率分布最终接近纯噪声(均匀分布)。
- $\mathbf{1}$ 代表一个 20 维的全 1 向量,对应于20 种氨基酸的均匀分布。
去噪过程(Denoising Process)
为了逆转上述前向扩散过程,并恢复抗体 CDR 序列,需要通过一个神经网络 $F(\cdot)$ 进行预测。该网络的输入是抗体-抗原复合体的上下文信息,输出是去噪后的残基分布:
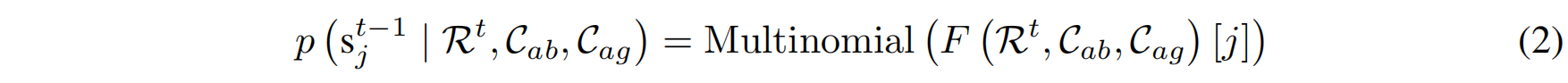
其中:
- $\mathcal{R}^t$ 是当前时间步 $t$ 的 CDR 片段。
- $C_{ab}$ 是抗体骨架(framework)。
- $C_{ag}$ 是抗原(antigen)。
- $F(\cdot)$ 是用于去噪的神经网络。
本文使用 DiffAb(Luo et al., 2022)作为生成模型的骨干(backbone),用于进行检索增强
4 Methods
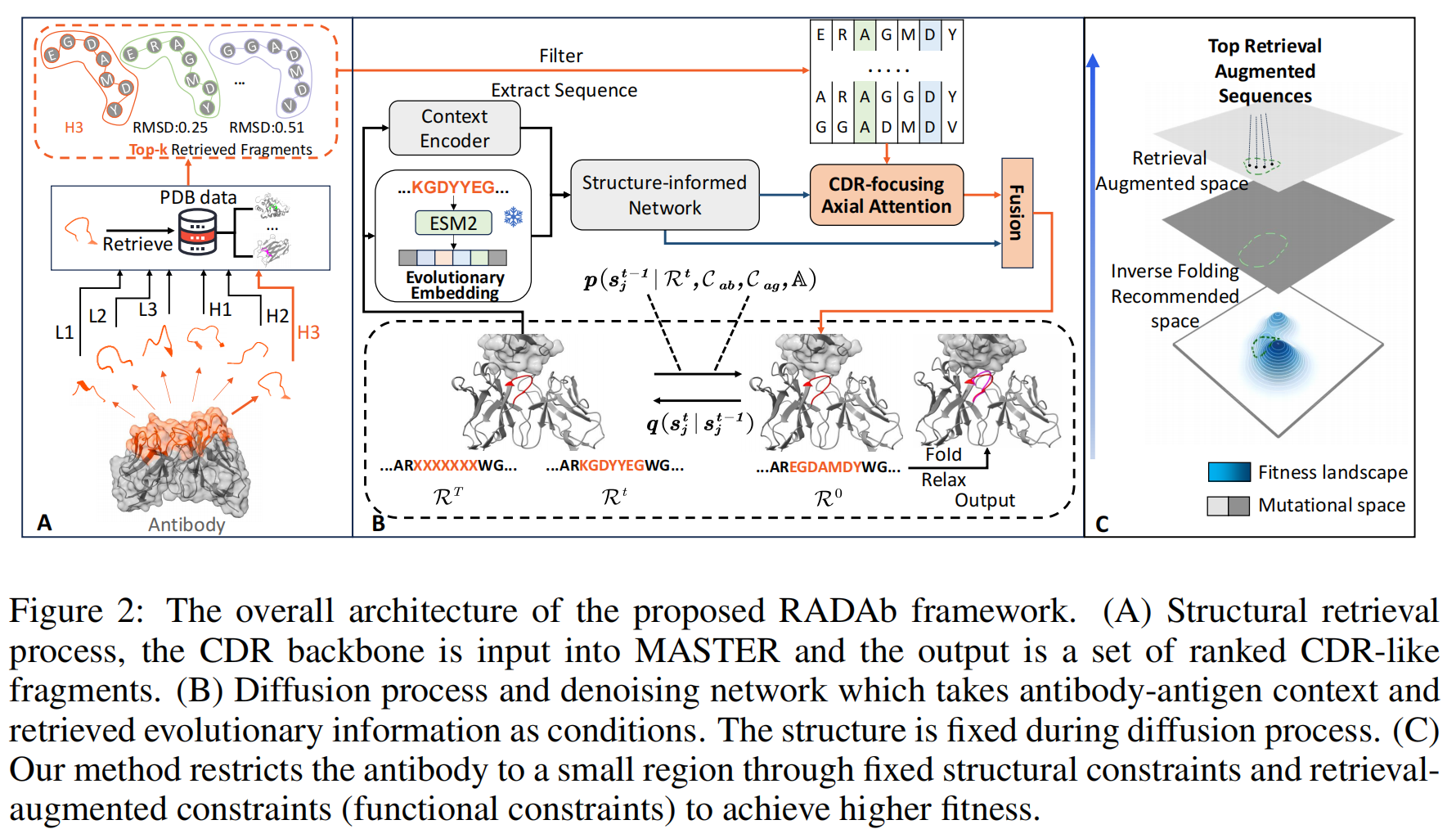
本研究提出 RADAb(如图 2 所示),这是一种基于结构信息的检索增强扩散框架,用于抗体序列设计和优化。该模型采用结构检索算法来搜索具有同源性的抗体结构,并将其序列作为扩散模型的条件输入,以提供同源模式和进化信息。
4.1 Structural Retrieval of CDR Fragments
蛋白质的结构由其序列决定,而能够折叠成相似结构的蛋白质序列通常具有相似的性质。因此,结构相似的蛋白质序列往往富含进化信息。基于这一原理,我们在 PDB(Protein Data Bank)数据库中进行检索,目标是找到与目标 CDR 结构相似且具有同源性的片段,并期望它们具有相似的功能。
检索方法:MASTER
为了平衡检索质量与速度,我们使用 MASTER(Zhou & Grigoryan, 2015)进行搜索。
MASTER 使用 主链原子(backbone atoms)的均方根偏差(RMSD) 作为相似性度量指标,能够高效地在 PDB 数据库中搜索与查询结构匹配的片段。
MASTER 的特点:
- 可以查询由一个或多个非连续片段组成的结构片段。
- 允许在给定的 RMSD 阈值 内找到所有匹配的片段。
- 仅利用蛋白质的 骨架信息(backbone information) 进行搜索,不会泄露序列数据。
检索过程
检索的具体过程如 Algorithm 1 所示,详细信息可见 附录 A.3。
- 计算 RMSD 评分并排序
- 对于检索到的 CDR 片段,我们计算它们与真实 CDR 片段的 RMSD,并以此进行排序。
- 之后,我们会过滤掉原始 CDR 片段,以确保训练时不会过度依赖原始数据。
- CDR 片段数据库
- 为了方便使用,我们进一步构建了一个CDR-like 片段数据库(详见附录 A.4)。
- 训练 vs. 生成
- 训练阶段:为了让模型学习更加丰富的进化信息,我们会去除完全相同的 CDR 片段,以防止模型过拟合。
- 生成阶段:我们不会执行相同的过滤,以提高生成质量。

4.2 Model Architectures
RADAb 模型利用抗原-抗体复合物的结构和序列上下文,结合检索得到的CDR-like 片段序列作为条件输入,进行逐步去噪生成抗体 CDR 序列。该模型由两个分支组成:
- 全局几何上下文信息分支(Global Geometry Context Information Branch)
- 负责学习抗原-抗体复合物的全局上下文信息。
- 局部 CDR 关注信息分支(Local CDR-Focused Information Branch)
- 负责学习 CDR-like 片段的局部同源信息,以捕获具有相似结构的残基间的功能相似性和进化信息。
这两个分支的输出共同用于生成最终的 CDR 序列。
4.2.1 Global Geometry Context Information Branch
(1)Context Encoder
蛋白质由多个残基连接形成,每个残基的特征主要包括:
- 单个残基:残基类型(Residue Type),主链原子坐标(Backbone Atom Coordinates),主链二面角(Backbone Dihedral Angles)
- 成对残基:残基类型对(Residue Pair Types),序列相对位置(Sequential Relative Position),空间距离(Spatial Distance),成对的主链二面角(Pairwise Backbone Dihedrals)
这些特征被拼接后,输入到两个独立的多层感知机(MLP),分别输出:$z_i$(单个残基特征),$y_{ij}$(成对残基特征)
(2)Evolutionary Encoder
近年来,基于结构信息的**蛋白质语言模型(PLM)**被证明是提取蛋白质序列嵌入和提供进化信息的优秀工具(Zheng et al., 2023; Shanker et al., 2024)。因此,我们选择 ESM2 作为抗体序列编码器。
具体而言,在时间步 t,将抗体序列(包含 CDR 区域)输入 ESM2,得到进化嵌入:$e^t = E(S_{ab} \cup R^t)$
- $S_{ab}$:抗体框架序列
- $R^t$:当前时间步 $t$ 的 CDR 序列
(3)Structure-Informed Network
- 以上编码结果作为条件输入,结合当前时间步的 CDR 序列和结构状态,输入到 一系列 Invariant Point Attention(IPA) 层(Jumper et al., 2021),转换为隐藏表示:$h_i = \text{IPA} (z_i, y_{ij}, e^t)$
- 随后,隐藏表示 $h_i$ 通过 MLP 转换为 CDR 位点的氨基酸类型概率分布:$r_{\text{global}} = \text{MLP}(h_i)$
- 该全局信息作为输入传递给局部 CDR 关注信息分支。
4.2.2 Local CDR-Focused Information Branch
(1)CDR-like 片段预处理
- 去除 CDR 片段,仅保留抗体框架序列。
- 填充 CDR-like 片段到抗体框架中,构造一个 CDR-like 片段矩阵 $E$。
(2)CDR-Focused Axial Attention
该分支由一组**轴向注意力层(Axial Attention Layers)**组成,被称为 CDR-Focused Axial Attention:
- CDR-like 片段结构与真实 CDR 结构高度相似,因此可利用 MSA-Transformer(Rao et al., 2021) 提出的**绑定行注意力(Tied Row Attention)**机制。
- 在标准轴向注意力(Axial Attention, Ho et al., 2019)中,行和列是独立计算的;
- 但在 MSA(Multiple Sequence Alignment) 中,各序列具有较高的结构相似性,我们的 CDR-like 片段矩阵也符合这一特性,因此:
- 计算某一行的注意力时,同时考虑其他行的注意力,提高模型对结构相似性的利用效率,同时降低显存占用。
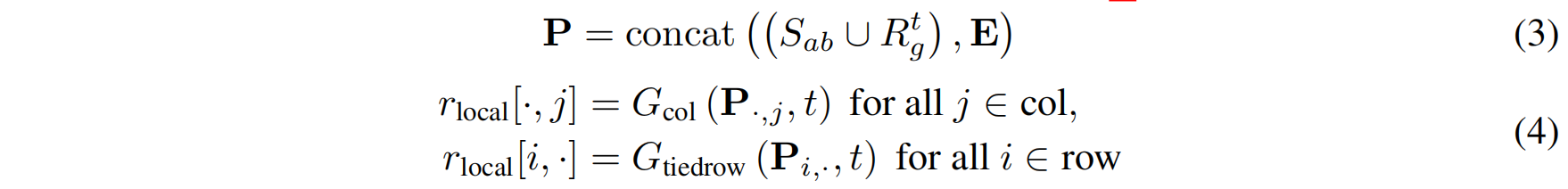
输入 CDR-Focused Axial Attention 的矩阵 P 构造公式3:
其中:
- 第 1 行:抗体框架序列 + 当前时间步的 CDR 噪声序列 $R^t$(从全局概率分布 $r_{\text{global}}$ 采样得到)。
- 第 2 - k 行:检索得到的CDR-like 片段矩阵 $E$(选取前 15 个相似片段)。
该矩阵输入 CDR-Focused Axial Attention 后,计算同源嵌入并得到 CDR 位点的局部概率分布:
- 列注意力(Column Self-Attention) 计算 CDR 残基与检索片段的关系。
- 行注意力(Row Self-Attention) 计算抗体-抗原序列内部的关系。
(3)Skip Connection for Information Fusion
虽然全局分支已生成 CDR 氨基酸的概率分布,但在前向传播过程中可能会丢失抗原-抗体上下文信息,公式4因此:
- 全局信息 $r_{\text{global}}$ 与 局部信息 $r_{\text{local}}$ 进行跳跃连接
- 通过 Softmax 计算最终 CDR 氨基酸概率分布。
4.3 Model Training and Inference
(1) The overall training objective
训练目标是使模型在每个时间步上预测的概率分布与真实后验分布尽可能接近。
采用 KL 散度(Kullback-Leibler Divergence) 作为损失函数:

训练过程中,对整个扩散过程的损失函数进行均匀采样:

(2) Conditional reverse diffusion process
从时间步 T 开始,将 CDR 位置初始化为均匀分布的噪声。
通过 ESM2 编码器 $E(\cdot)$、全局上下文网络 $F(\cdot)[j]$ 和局部 CDR 网络 $G(\cdot)[j]$,预测每个时间步的噪声分布并进行去噪:
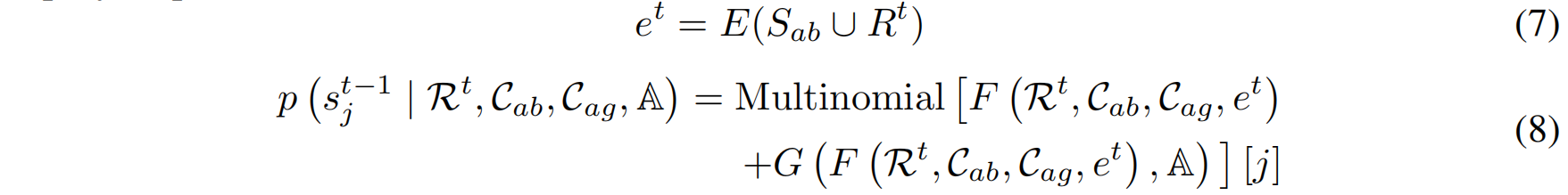
采样时,移除 CDR 片段,填充随机噪声,并利用检索得到的 CDR 片段来引导扩散模型进行逐步去噪,最终生成完整的 CDR 序列。
5 Experiments
为了评估模型的生成能力,设定了两个主要任务:
- 抗体 CDR 序列逆向折叠(Antibody CDR Sequence Inverse Folding)(5.1),即在已知抗体骨架结构的情况下,生成对应的 CDR 序列。
- 基于序列设计的抗体优化(Antibody Optimization Based on Sequence Design)(5.2),用于优化抗体序列以提高其亲和力或稳定性。
此外,还进行了一系列消融实验(5.3),以验证检索增强方法的有效性。
数据集与预处理
- 训练数据来自 SAbDab 数据库,并结合了我们构建的 CDR 类似片段数据集。
- 参照 Luo et al., 2022 的处理方法:
- 移除分辨率低于 4Å 的结构。
- 排除靶标为非蛋白质抗原的抗体。
- 使用 ANARCI(Dunbar & Deane, 2016)进行抗体残基编号(基于 Chothia & Lesk, 1987)。
- 以 CDR-H3 50% 序列相似性进行聚类,并选择 **50 个 PDB 文件(共 63 组抗原-抗体复合物)**作为测试集。
- 确保测试集与训练集无重叠(即移除训练集中与测试集同属一个簇的结构)。
5.1 Antibody CDR Sequence Inverse Folding
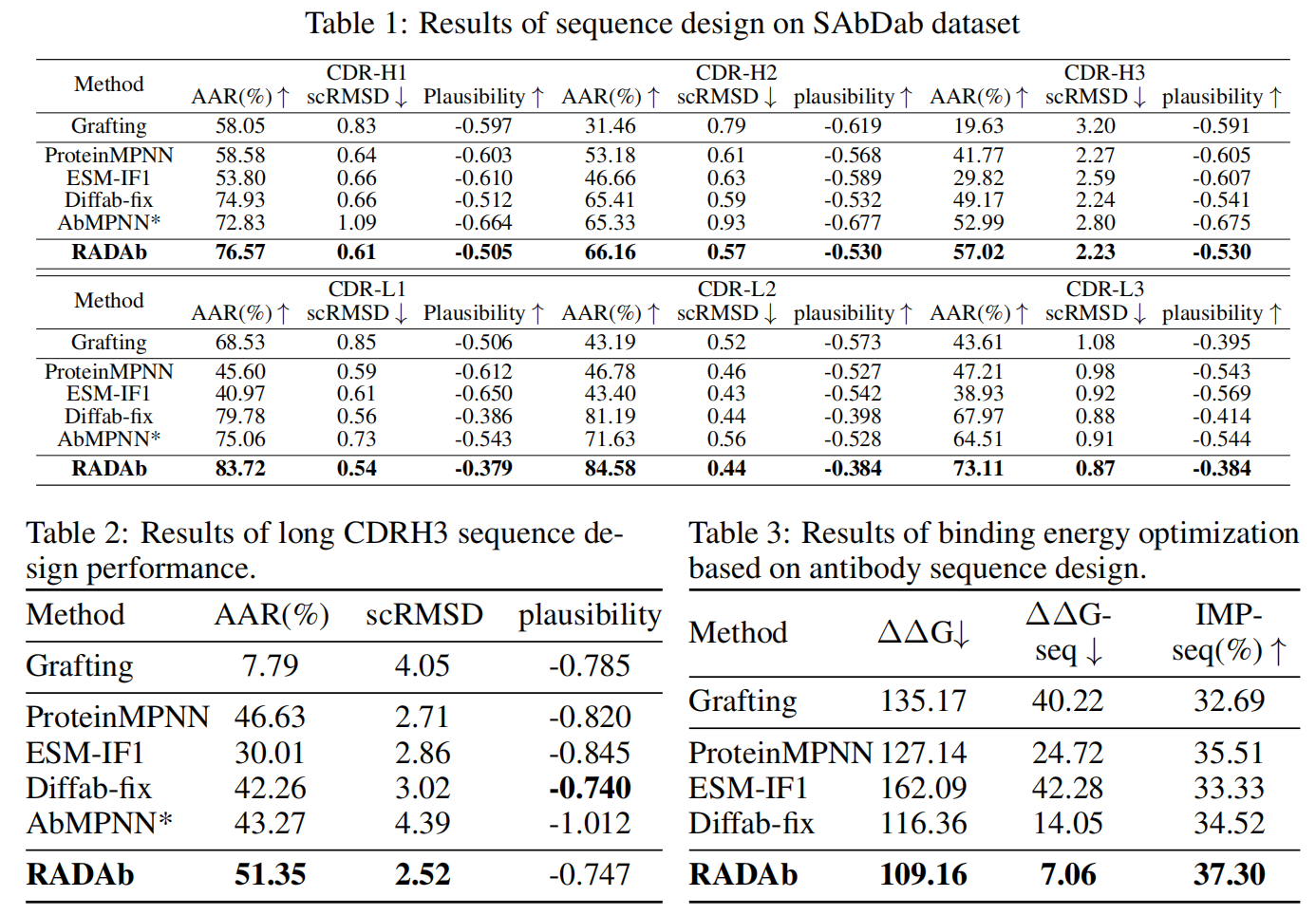
实验结果见 Table 1:
- RADAb 在 所有指标和所有 CDR 区域上均优于 现有的最先进方法。
- 特别是在 CDR-H3(最具变异性和特异性的区域),RADAb在 AAR 指标上相较于 Diffab-fix 和 AbMPNN 提升明显。
- 结果表明:检索增强方法 通过引入 结构相似的同源序列,有效提高了模型的 准确性、一致性和合理性。
此外,CDR-H3 由于 长度、序列和结构变异性大,通常 深度学习模型在生成较长 CDR-H3 序列时表现较差(Luo et al., 2022; Høie et al., 2024)。因此,我们 从测试集中选取 CDR-H3 长度大于 14 的子集 进行额外评估,结果见 Table 2:
所有方法的生成性能均有所下降,但RADAb依然保持 较强的一致性,并且 相较于其他方法的改进幅度更大。
5.2 Antibody Functionality Optimization
在本节中,关注抗体序列的进化,并评估进化后的序列在折叠后的三维结构中是否比原始抗体序列具有更好的功能性(即更强的抗原结合能力)。
- 折叠设计的 CDR-H3 序列:
- 通过 ABodyBuilder2 将生成的 CDR-H3 序列 与抗体框架序列结合,并预测完整的蛋白质结构。
- 同时,将 原始真实抗体序列 也折叠成完整蛋白质结构,作为对照组。
- 能量优化与计算:
- 使用 PyRosetta(Alford et al., 2017)中的 FastRelax 和 InterfaceAnalyzer:
- FastRelax:对抗原-抗体复合物进行结构松弛(Relaxation)。
- InterfaceAnalyzer:计算抗体-抗原复合物的结合能(Binding Energy, ∆G),用于评估抗体的结合能力。
- 使用 PyRosetta(Alford et al., 2017)中的 FastRelax 和 InterfaceAnalyzer:
Table 3 结果分析:
- RADAb设计的抗体序列,在折叠和结构松弛后,结合能显著降低,比其他方法更稳定。
- 其中 37.3% 的设计序列,其结合能低于原始抗体序列折叠后的结合能,表明序列优化在一定比例上成功改善了抗体的结合功能。
特定抗体优化案例分析:
- 我们从测试集中选择了一个抗原-抗体复合物(靶向 SARS-CoV-2 受体结合域的中和抗体,PDB: 7d6i)。
- 生成 50 个 CDR-H3 序列 并计算其折叠后的结合能(∆G)。
- 其中 68% 的样本比原始复合物具有更低的结合能,即优化后的序列表现出更强的抗原结合能力。
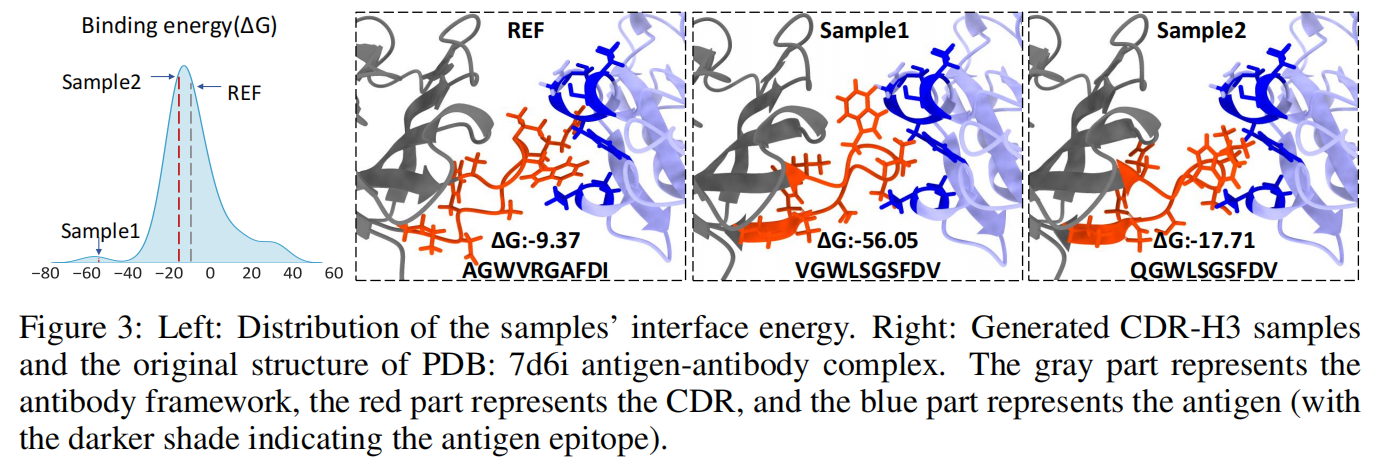
Figure 3 结果分析:
- 选取了两个具有代表性的样本:
- 尽管它们没有获得最高的氨基酸恢复率(AAR),但它们的结合亲和力显著优于原始抗体结构。
- 这表明:优化后的抗体序列可能会发生突变,导致部分氨基酸序列不同于原始抗体,但整体结构更有利于抗原结合。
5.3 Analysis
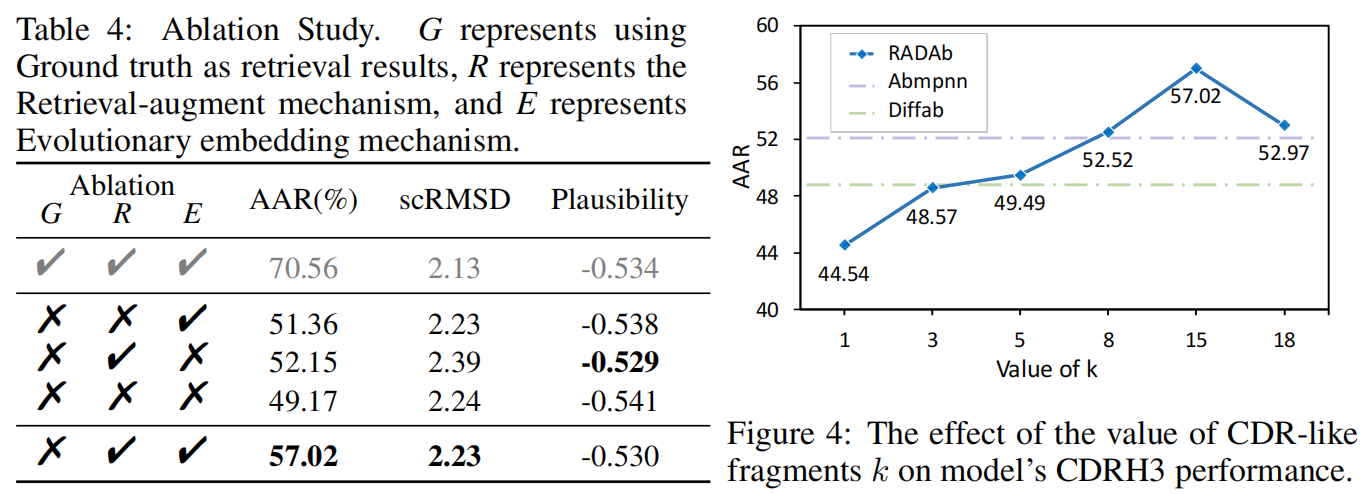
实验结果(Table 4)
- 首先用 CDR 序列的真实值(ground truth)作为输入,验证检索增强模块的效果。
- 分别去除检索增强机制和进化嵌入机制,评估它们的单独贡献。
- 实验结果表明:
- 检索增强模块和进化嵌入模块单独使用时,都能提升模型的性能。
- 两者结合使用时,模型性能达到最优。
- 这表明,检索增强和进化嵌入是互补的,能够协同提升抗体序列的生成质量。
实验结果(Figure 4)
- 当 k 过低时(例如 k = 1 或 2),模型的性能下降,甚至可能带来负面影响。
- 可能的原因:检索数据不足,导致模型过拟合(overfitting)。
- 随着 k 的增加,模型的性能逐步提高,表明检索到的 CDR 片段为模型提供了有益的信息。
- 当 k = 15 时,模型达到最佳性能。
- 当 k > 15 后,性能开始下降,可能是因为过多的检索片段引入了噪声,影响了模型的泛化能力。

