论文地址:Prot42: a Novel Family of Protein Language Models for Target-aware Protein Binder Generation
Prot42:预训练GPT设计binder
Abstract
本论文针对蛋白质工程的一个核心难题:传统方法复杂、耗时且资源密集。当前的生成式 AI 技术虽然有突破,但大多依赖目标蛋白的三维结构和特定结合位点信息,例如 AlphaProteo 和 RFdiffusion。这些模型在缺乏结构信息的情况下会面临严重瓶颈。
为了突破这一限制,作者提出了 Prot42 ——一个全新的 Protein Language Model (pLM) 家族。该模型以大规模的无标注蛋白序列进行预训练,通过 先进的自回归解码器结构(受自然语言处理中 decoder-only 模型的启发),捕捉蛋白质的进化、结构和功能特征。
Prot42 模型的突出特点之一是:它支持输入长达 8192 个氨基酸残基的序列,这远超当前主流模型(如 ProtBert、ESM-1b 通常限制在 1024 个残基左右)。这使得 Prot42 能够建模大型蛋白和多结构域序列,适用于更复杂的生物分子场景。
此外,论文展示了两个实际应用场景:
- 高亲和力的蛋白质结合体(protein binders)生成
- 序列特异性的 DNA 结合蛋白生成
模型可在 huggingface 上公开获取,作为一个高效、精确、可扩展的工具包,支持快速的蛋白质工程任务。
1 Introduction
蛋白质结合体(protein binders),包括抗体和工程蛋白,在生物技术和治疗应用中起着关键作用,涉及从诊断到靶向药物输送等多个领域。传统的生成特异性结合体的实验方法存在资源消耗大和组合复杂性限制的问题。近年来,结构驱动的计算方法成为一种有前景的替代方案,例如 AlphaProteo 和 RFdiffusion,这些方法在实验中的成功率更高,所需筛选的候选数量也显著减少。尽管如此,这些方法仍存在关键局限:它们依赖目标蛋白的三维结构以及明确的结合位点信息,这对那些结构信息缺乏的目标蛋白构成了瓶颈。
蛋白语言模型(Protein Language Models, pLMs)提供了另一种可行途径,它们主要在序列层面工作,利用大规模无标注蛋白序列数据学习进化关系和结构属性的表示。已有研究表明,pLMs 在蛋白的功能注释、结构预测和新序列生成等任务上相较于传统的基于物理化学特征或统计特征的方法有显著优势。然而,目前的 pLMs 存在两大限制:一是最大可处理的序列长度有限,二是缺乏原生的生成能力。例如,ESM-1b 和 ProtBert 等模型虽然在表示学习方面表现优异,但不能生成新序列,且通常只能处理最多约 1024 个氨基酸,这使得它们在处理复杂蛋白质和结合界面建模任务时效果受限。虽然像 Evo-2 这样的基础模型支持上百万 token 的上下文窗口,但它主要面向 DNA 建模任务,并未解决蛋白质层面的生成问题,因此仍存在对专门的蛋白生成模型的需求。
在本文中,我们提出了 Prot42 ——一个新的 蛋白语言模型家族,采用自回归的 decoder-only 架构,灵感来自于 NLP 中如 LLaMA 模型的最新进展。我们预训练了两个 Prot42 变体,分别拥有 5 亿和 11 亿参数,起初支持最多 1024 个氨基酸残基。通过持续预训练(continuous pretraining),我们将上下文长度扩展至 8192,从而使模型能够捕捉到对建模大型蛋白质、多结构域复合体和复杂分子相互作用至关重要的长距离依赖关系。这种增强的表示能力对于生成高亲和力的蛋白结合体非常关键,也有助于发现新的生物分子相互作用。
我们详细描述了 Prot42 的模型架构、预训练策略,以及上下文长度扩展机制,并展示了其在序列建模精度方面的提升。我们使用困惑度(perplexity)*作为评估指标,验证 Prot42 在更长序列上的预测性能。为了验证其实用性,我们重点展示了两个实际应用:**蛋白结合体生成**和*序列特异性的 DNA 结合蛋白生成。结果表明,Prot42 拥有先进的生成能力**,能够推动计算蛋白设计的边界,实现快速、精准和可扩展的蛋白质工程。
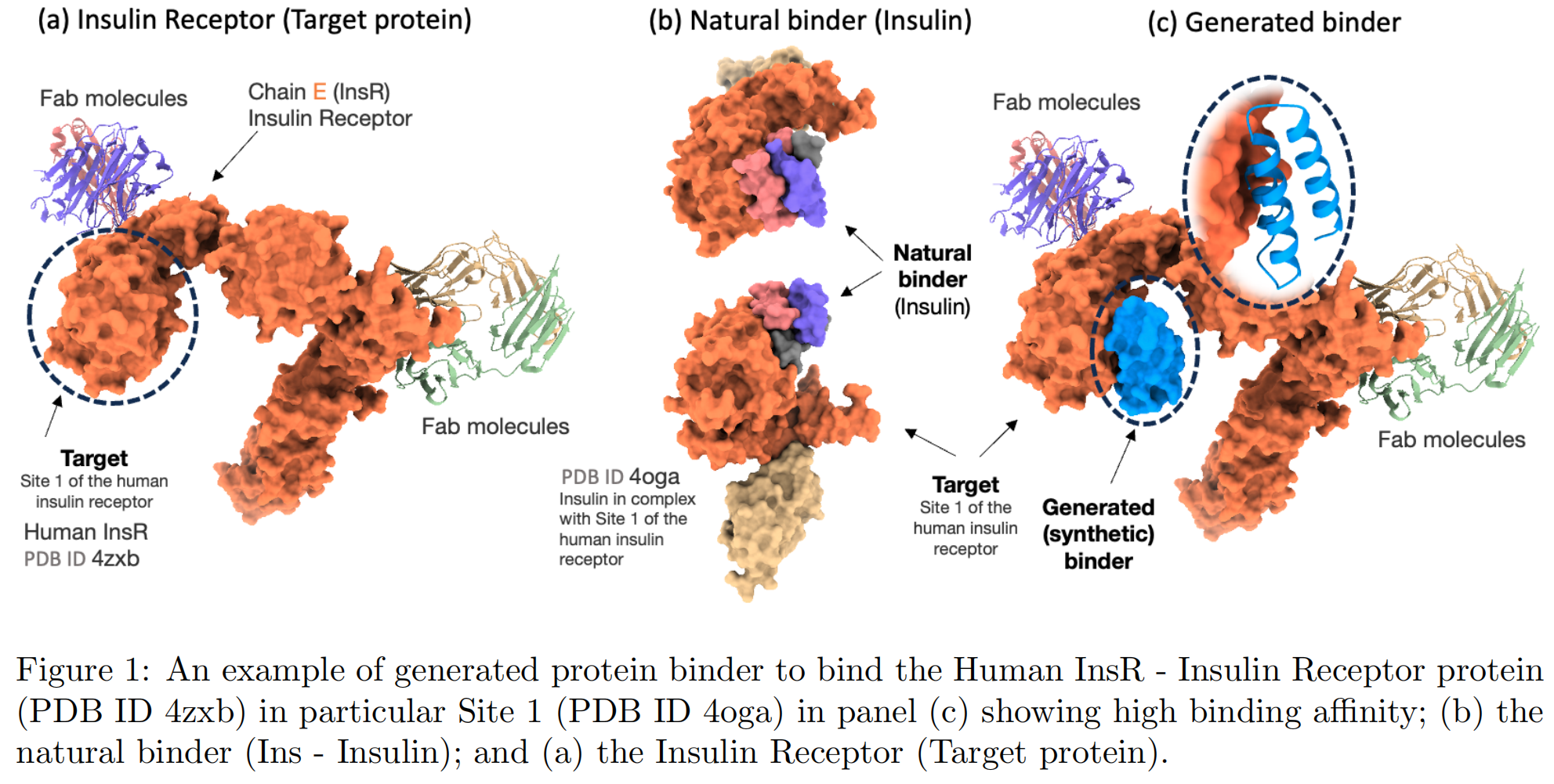
作为 Prot42 的能力展示,我们在图 1 中给出一个示例:模型生成了一个高亲和力的结合体,靶向的是胰岛素受体(InsR)α 亚基,并与天然结合体 胰岛素(PDB ID 4oga)进行了比较。
3 Methodology
本节展示了如何使用 Prot42 首次实现一种 instruction-tuning(指令微调)的方法,用于从目标蛋白序列直接生成高亲和力、长序列的蛋白结合体,完全不依赖结构信息或额外的辅助任务训练目标。
通过一种新颖的多模态策略整合了 Gene42 的基因组嵌入 和 Prot42 的蛋白质嵌入,进一步扩展模型能力以生成 序列特异性的 DNA 结合蛋白。这一方法消除了对明确结构限制或**预定义结合基序(motifs)**的依赖,强调了计算模型在设计具有功能的蛋白结合体时对多种分子上下文的适应能力。
3.1 Data Preparation
使用了 UniRef50 数据集,包含 6320 万条氨基酸序列,并使用一个由 20种标准氨基酸组成的词表进行分词。为处理不常见或模糊的残基,引入了 X token,表示任意氨基酸。
每条序列都被限制在最大长度 1024 tokens,超出长度的序列被剔除。最终我们得到了一个筛选后的子集,包含 5710 万条序列,初始的token 填充率(packing density)为 27%。为了更好地利用计算资源并提升效率,采用了 可变序列长度(VSL)打包技术,使得在固定上下文窗口中最大化 token 的占用。
这种方法将数据集减少到 1620 万条打包后的序列,同时实现了 96% 的填充效率,在保持序列多样性和完整性的同时显著提升了计算效率
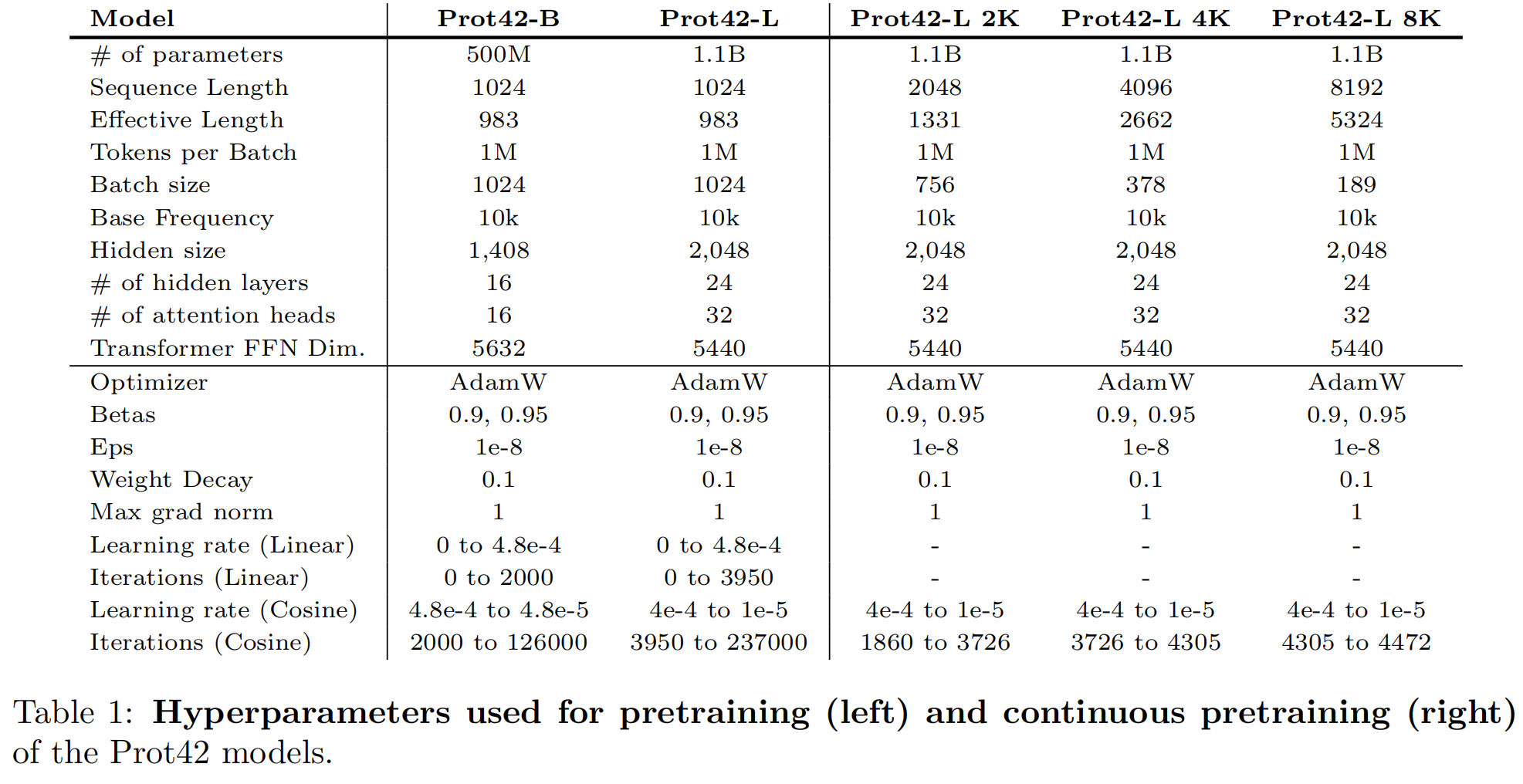
训练了以下模型变体,参数配置如下
3.2 Model Architecture and Pretraining
Prot42 是一个 基于 LLaMA 的 decoder-only 自回归 Transformer 模型。训练了两个主模型:Prot42-B:5亿参数,Prot42-L:11亿参数
使用了一种 maximal update parametrization (μP) 的超参数选择方法。具体流程是:先训练一个 81M 参数的小模型,用于超参数搜索;再将这些超参数通过 μ-transfer 转移到 500M 和 1.1B 模型中。
训练期间,每个 batch 的总 token 数固定为 100 万,学习率使用 线性 warm-up + cosine 衰减策略,并全部在 Cerebras CS-2 上进行训练。
Continuous Pretraining for a Larger Context Length
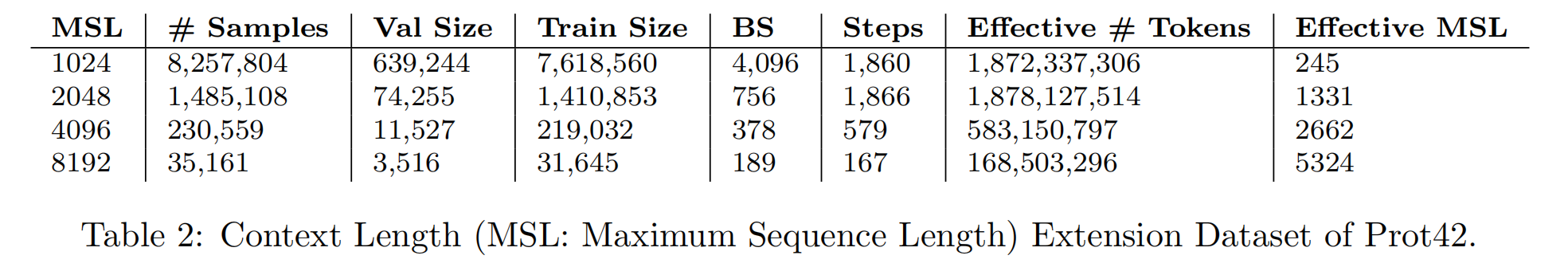
将 Prot42-L 从支持 1024 序列长度的版本,持续扩展至支持 8192 残基,通过 **逐步增加最大序列长度(MSL)**实现。
训练流程:
- 前10%的训练步骤使用 MSL = 1024;
- 随后阶段使用逐步增加的 MSL:2048 → 4096 → 8192;
- 每阶段保持有效 token 数恒定为 1M。
数据划分见 Table 2。每个阶段的 MSL、样本数量、步数、平均有效长度等均有详细记录。最终训练阶段实现了最高 5324 的有效序列长度。
Model Evaluation using Validation Perplexity
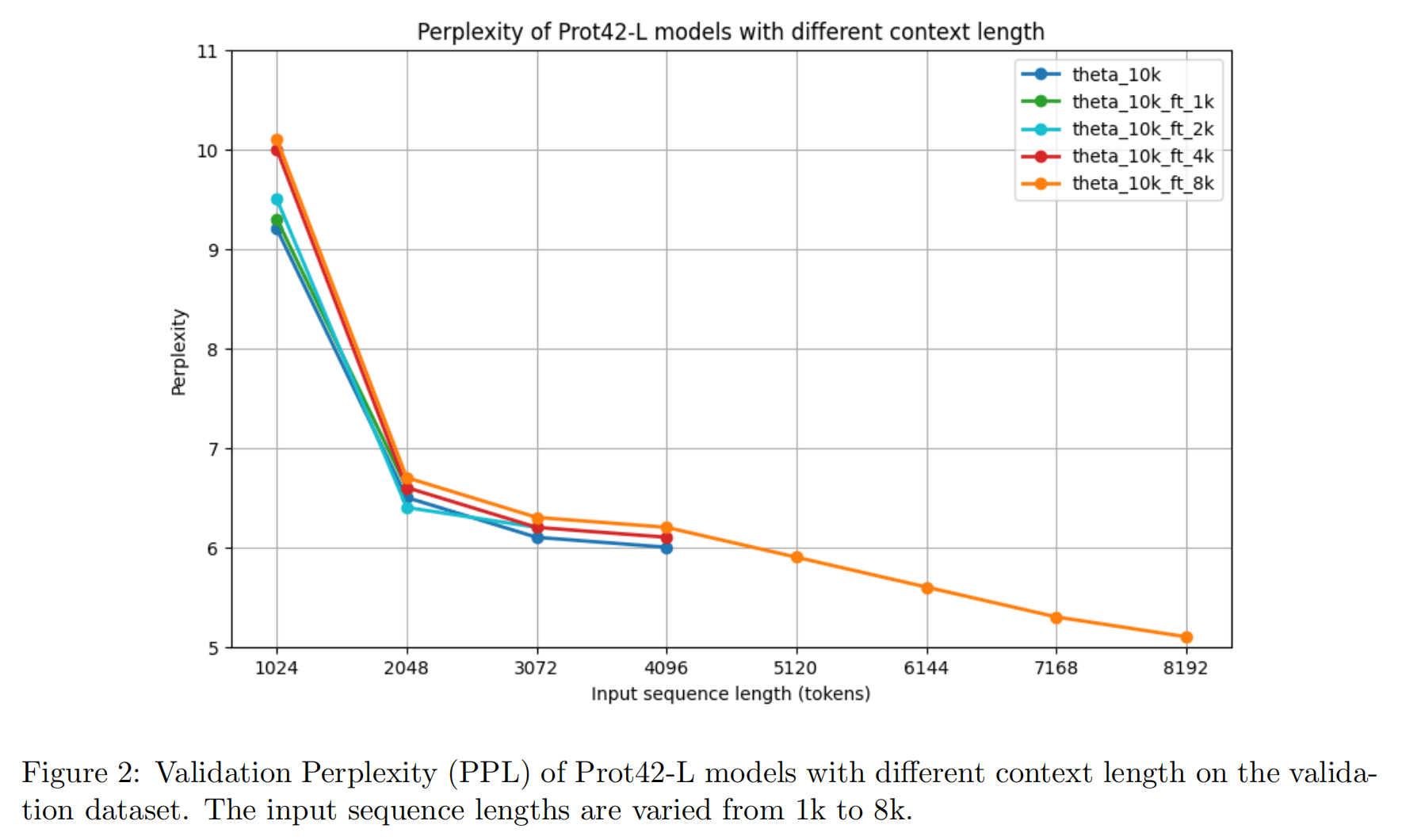
使用 perplexity(PPL) 作为评估自回归模型性能的标准指标。对不同上下文长度的模型(1K 到 8K)在验证集上进行评估。结果显示:
- 所有模型在 1024 tokens 时 PPL 较高(9–10);
- 在 2048 tokens 降至约 6.5;
- 8K 模型在最大上下文长度下 PPL 降至最低的 5.1,表明它能更好地建模长距离依赖关系;
- 特别是 4096–8192 token 区间的持续下降,显示其优越的长序列建模能力。
Embeddings Evaluation
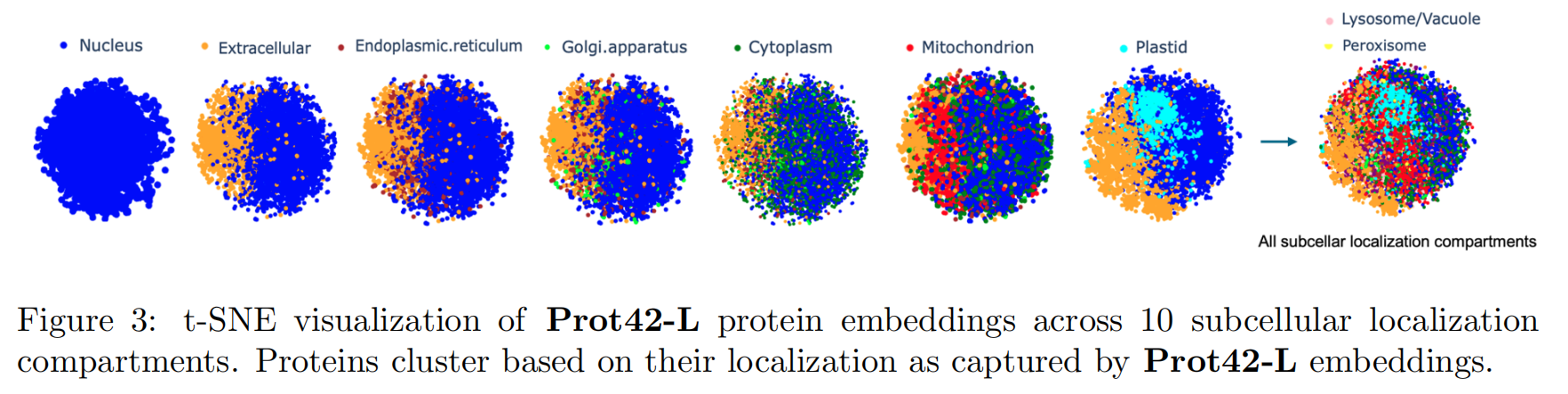
使用来自 PEER benchmark + UniProt 注释数据的蛋白质序列,用于预测 10 类亚细胞定位(如细胞核、线粒体、质膜等)。
方法:将每个蛋白嵌入为大小为 32×2048 的高维向量,对其进行平均池化处理以获得全局表示。再使用 t-SNE 降维可视化,显示模型能清晰地区分不同亚细胞定位的蛋白类别。
结果表明:Prot42-L 学到的嵌入具有生物学意义,可用于下游任务如蛋白功能预测、药物靶点发现、合成生物设计等。
3.3 Protein Binder Generation
蛋白质通常不会孤立地发挥作用,而是通过相互作用、结合形成复杂网络来驱动细胞过程。设计能够特异性结合目标蛋白的蛋白质,是分子生物学中的一个基本问题,具有广泛的应用,包括治疗、合成生物学等方向。
设目标蛋白的氨基酸序列表示为 $x = (x_1, x_2, …, x_n) \in \mathcal{X}$,其中 $x_i$ 表示目标蛋白第 $i$ 个残基;设结合体蛋白序列为 $y = (y_1, y_2, …, y_m) \in \mathcal{Y}$,表示能够与目标蛋白结合的序列。目标是建模条件概率分布 $p(y | x)$,即在给定目标蛋白序列 $x$ 的前提下,生成结合体序列 $y$ 的概率分布。
为此,作者使用了一个受机器翻译任务启发的序列到序列(sequence-to-sequence)模型。该模型在训练时使用一个由蛋白互作对组成的数据集 $\mathcal{B} = {(x_i, y_i)}$,其中每对 $(x, y) \in \mathcal{B}$ 被拼接为:$s = (x_1, …, x_n, \texttt{[SEP]}, y_1, …, y_m)$。其中 [SEP] 是一个特殊分隔符 token,用于标识目标序列与结合体序列的分界。
训练过程中,模型优化以下自回归损失函数:
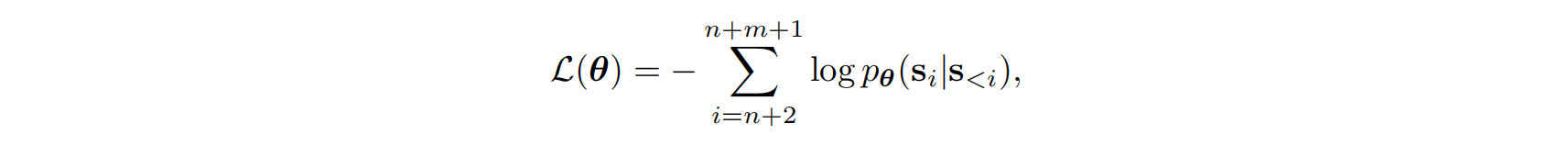
其中 θ 表示模型参数,损失函数从目标序列末尾加上分隔符后的第一个 token 开始,逐步学习生成整个结合体序列。
在推理(inference)时,模型以目标蛋白序列 x 为条件,接上 [SEP] token 以及一个起始的甲硫氨酸残基(“M”)作为初始输入。训练集中所有蛋白序列通常以甲硫氨酸开始,因此使用 “M” 可以有效引导生成。
结合体序列 y 的生成过程是逐步自回归的,满足:
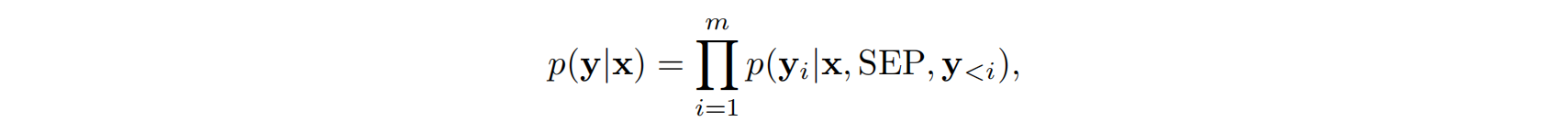
确保生成序列的多样性和质量,作者使用了随机采样策略,结合了温度控制(temperature scaling)、nucleus sampling(top-p)和 top-k 筛选。具体地,对于第 i 个 token,其被采样的概率为:
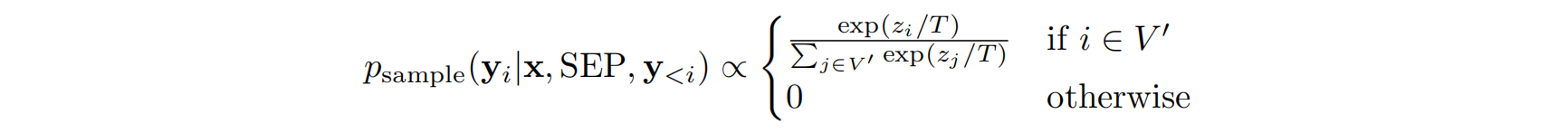
其中 T 是温度参数,控制生成的随机性;$z_i$ 是当前 token 的 logit 值;$V’ \subseteq V$ 是在 top-k 和 top-p 限制下保留的候选 token 子集。
整个生成过程可表示为:
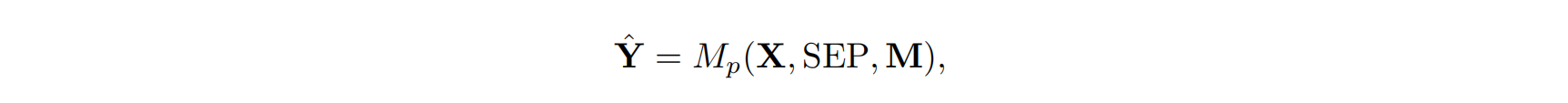
其中 $\hat{Y} = (y_1, y_2, …, y_m)$ 是生成的结合体序列,$M_p$ 表示 Prot42 模型。
图 4 提供了一个实际示例:目标蛋白为 VEGF-A(血管内皮生长因子 A),其链 V 被用作结合目标。模型学习了 $p(y \mid x)$ 并自回归地生成 $\hat{Y}$,输出结合体蛋白。
在推理阶段,模型以 VEGF-A 序列为输入,以 [SEP] 和 “M” 启动解码过程,然后按照上述采样策略生成每一个氨基酸残基。
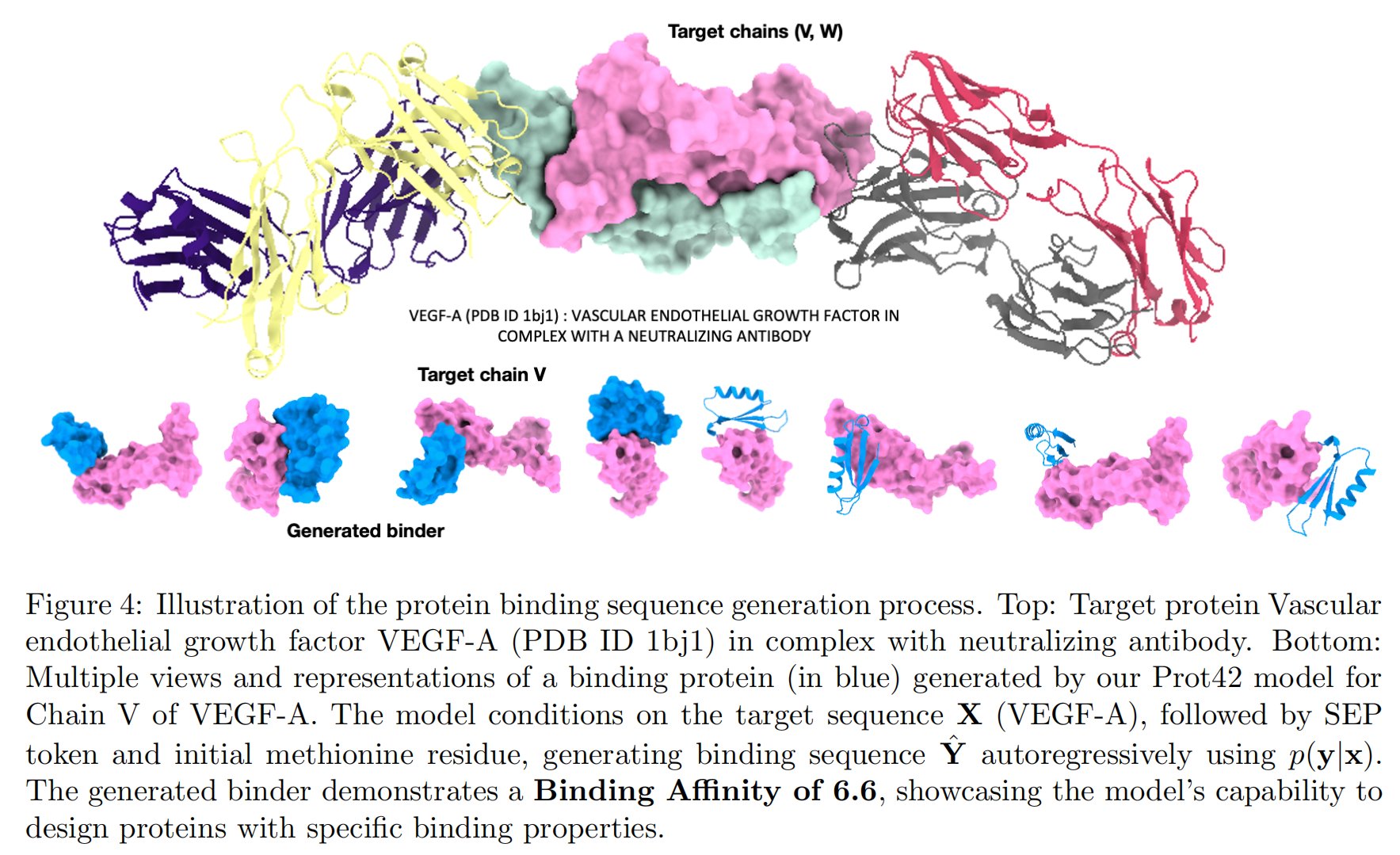
结构层面,图中显示了生成的结合体与目标蛋白之间的结合区域,并计算了它们之间的结合亲和力 $A(\hat{Y}, X)$,用于定量评估生成效果。
3.4 DNA Sequence-specific Binders Generation
除了蛋白-蛋白相互作用,设计能与特定 DNA 序列结合的蛋白质同样具有重要意义,这在基因调控和基因组工程领域开辟了新的前沿方向。
设有一个 DNA-蛋白序列对的数据集 $\mathcal{D}$,目标是生成一个蛋白序列 $\hat{X}$,其能有效结合某个给定的 DNA 序列。具体而言,一个 DNA-蛋白对可表示为三元组 $(X_d^{(1)}, X_d^{(2)}, X_p) \in \mathcal{D}$,其中:
- $X_d^{(1)} = (d_1^{(1)}, d_2^{(1)}, …, d_n^{(1)})$和 $X_d^{(2)} = (d_1^{(2)}, d_2^{(2)}, …, d_n^{(2)})$是长度为 $n$ 的两条 DNA 链,属于某个 DNA 词表 $\mathcal{V}_d$,分别表示正负链。
- $X_p = (p_1, p_2, …, p_m)$ 是由 $m$ 个氨基酸 token 构成的蛋白质序列。
DNA 模型 $M_d$ 将 DNA 序列编码为潜在表示:
- $H_d^{(1)} = (h_1^{(1)}, …, h_n^{(1)})$,来自 $X_d^{(1)}$
- $H_d^{(2)} = (h_1^{(2)}, …, h_n^{(2)})$,来自 $X_d^{(2)}$ 其中每个 $h_t^{(i)} \in \mathbb{R}^{1408}$ 是 DNA 模型输出的嵌入向量。
蛋白模型 $M_p$ 将蛋白序列 $X_p$ 编码为表示序列:$E_p = (e_1, e_2, …, e_m), \quad e_i \in \mathbb{R}^{2048}$
为将 DNA 上下文整合进蛋白质生成,作者对 DNA 表示做了投影,将其维度从 1408 映射至 2048:
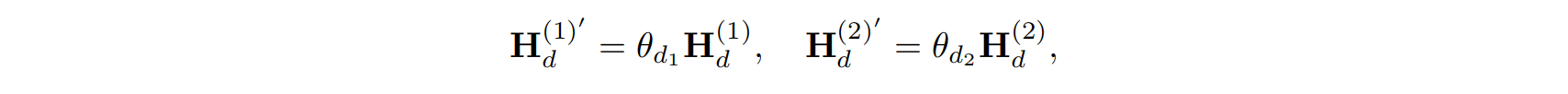
整合机制采用交叉注意力(cross-attention),用于将 DNA 信息与蛋白嵌入融合。注意力分数计算如下:
其中:$A = \text{Softmax}\left(\frac{QK^\top}{\sqrt{2048}}\right)$
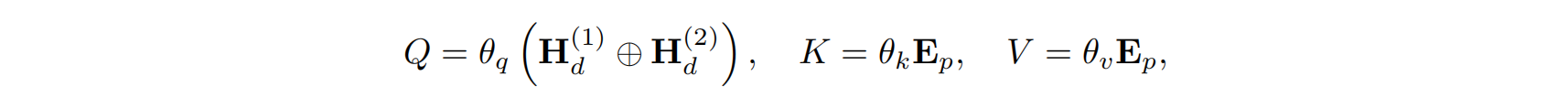
- 查询向量 $Q = \theta_q (H_d^{(1)} \oplus H_d^{(2)})$
- 键和值向量 $K = \theta_k E_p$, $V = \theta_v E_p$
这里 $\oplus$ 表示拼接操作,$\theta_q, \theta_k, \theta_v \in \mathbb{R}^{2048 \times 2048}$ 是可训练的参数。
由此得到 DNA 调制后的蛋白表示:$C_p = A V, \quad E_p’ = E_p + C_p$
其中 $C_p$ 是融合后的上下文信息,$E_p’$ 是最终输入到 protein decoder 的特征。
蛋白质生成仍然遵循自回归机制:每个位置的 token 由之前生成的 token 以及 DNA 上下文决定。输出概率分布如下:$p_t = \text{Softmax}(\theta_h e_t’ + \theta_c C_t), \quad \hat{p}t = \arg\max_j (p{t,j})$
其中 $\theta_h, \theta_c$ 是可学习参数,最终的蛋白质序列为:
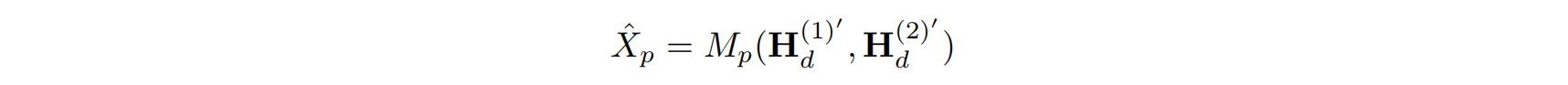
在实际推理中,先将两条 DNA 链编码成嵌入表示,传入蛋白模型 $M_p$。然后,以一个固定的起始 token(“M”)作为初始氨基酸残基,逐步生成后续序列。通过固定起始 token,模型可以专注于结合位点的预测生成。
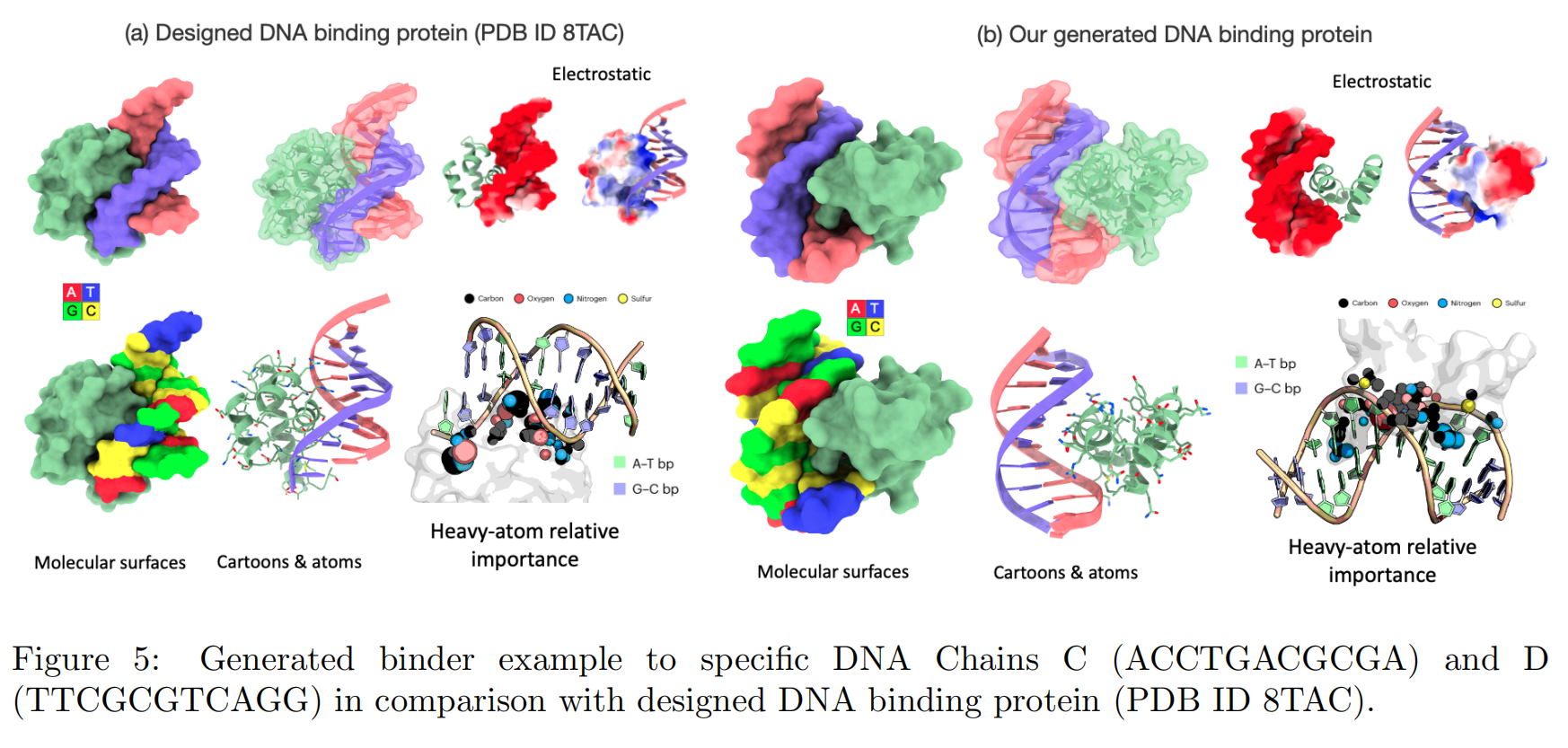
图 5 展示了一个具体示例,其中 DNA 链 C = ACCTGACGCGA,D = TTCGCGTCAGG,Prot42 根据 DNA 信息生成了与之结合的蛋白序列,并与已知 DNA 结合蛋白(PDB ID: 8TAC)进行比较。可视化结构显示模型能在空间结构上有效实现 DNA-蛋白的匹配。
4 Experimental Evaluation
4.1 Evaluation on PEER Benchmarks
在 PEER benchmark(Protein sEquence undERstanding)上,Prot42 被用于 14 项下游任务,涵盖:
- 蛋白功能预测(如荧光、稳定性、酶活性、溶解性)
- 亚细胞定位预测
- 结构预测(如接触图、折叠分类、二级结构)
- 蛋白-蛋白/小分子相互作用预测
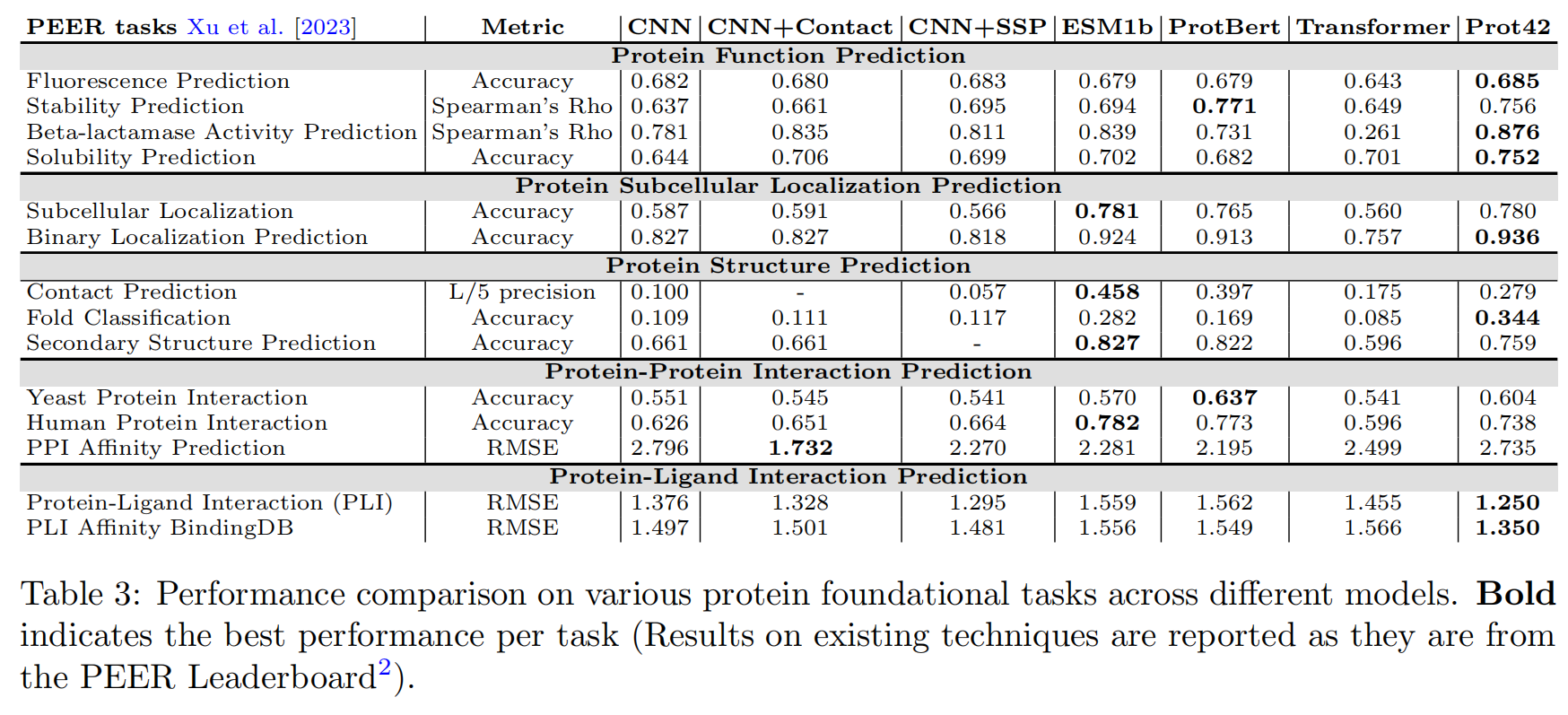
结果总结:
- Prot42 在 功能预测任务中表现优异,尤其在稳定性、酶活性和溶解性方面达到最优。
- 在 亚细胞定位任务中,其准确率与 ESM-1b 等模型持平或略优。
- 在 结构相关任务中(如接触图、二级结构),Prot42 结果具有竞争力。
- 在 蛋白-蛋白/蛋白-小分子相互作用预测中,也取得了可靠结果。
此外,在蛋白-配体任务中,作者整合了 Chem42 生成的化学嵌入,进一步提升预测性能。
4.2 Protein Binders generation
该任务评估 Prot42 相较于结构依赖方法(如 AlphaProteo)在设计结合蛋白方面的能力。
目标蛋白包含:BHRF1(EB 病毒抗凋亡蛋白)、SARS-CoV-2 RBD、IL-7Rα(白细胞介素受体)、PD-L1(免疫检查点)、TrkA(神经营养因子受体)、IL-17A(炎症因子)、TNF-α(肿瘤坏死因子)
实验流程:
- 使用 STRING 数据库中的高置信度互作对(score ≥ 90%)训练模型;
- 限制目标与结合体序列长度不超过 250;
- 微调后,针对每个目标生成 500 个候选结合体;
- 使用 Boltz-1 预测目标-结合体复合结构;
- 使用 PRODIGY 工具评估:结合自由能(ΔG)、解离常数(Kd)、界面面积、接触残基数、极性分布等指标
核心结果:
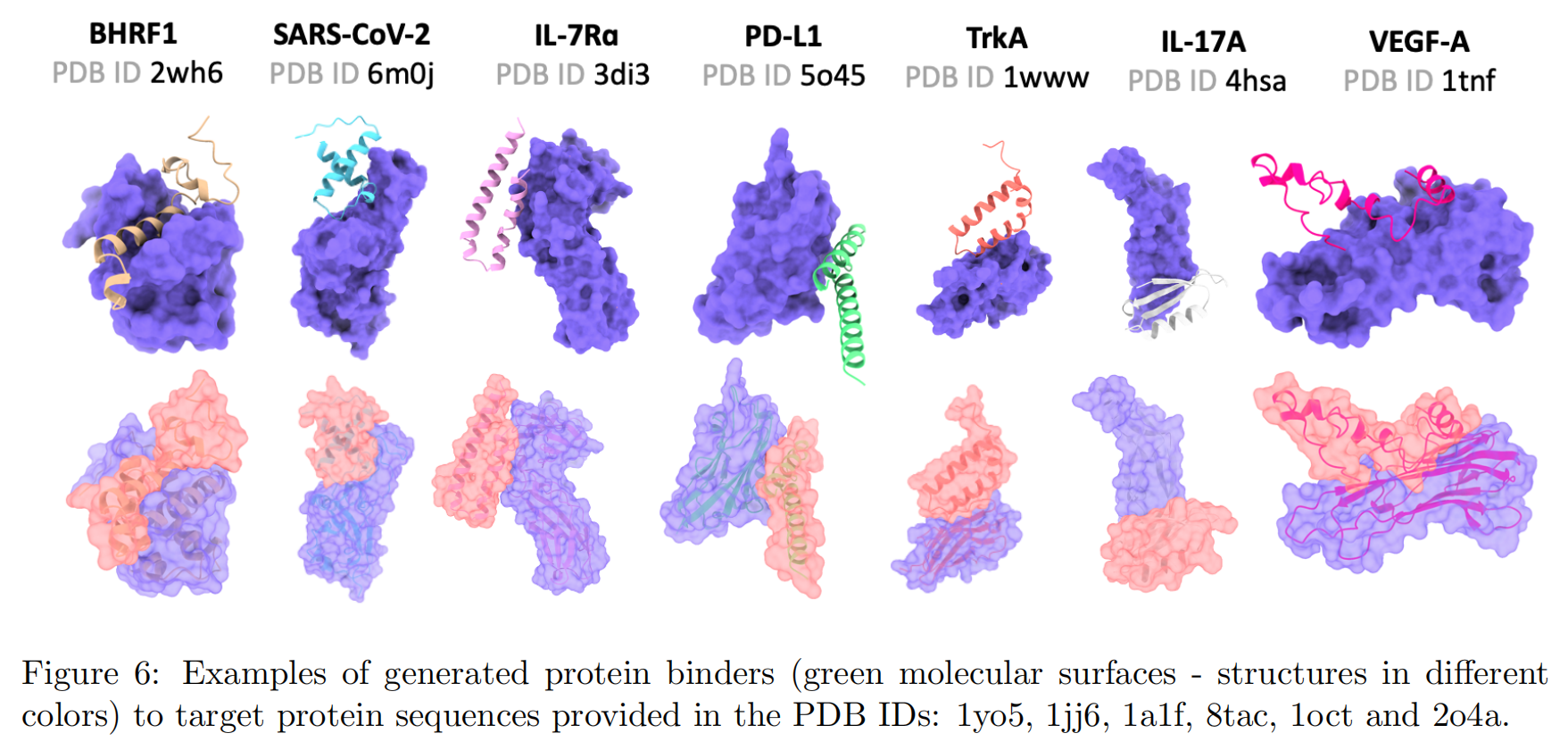
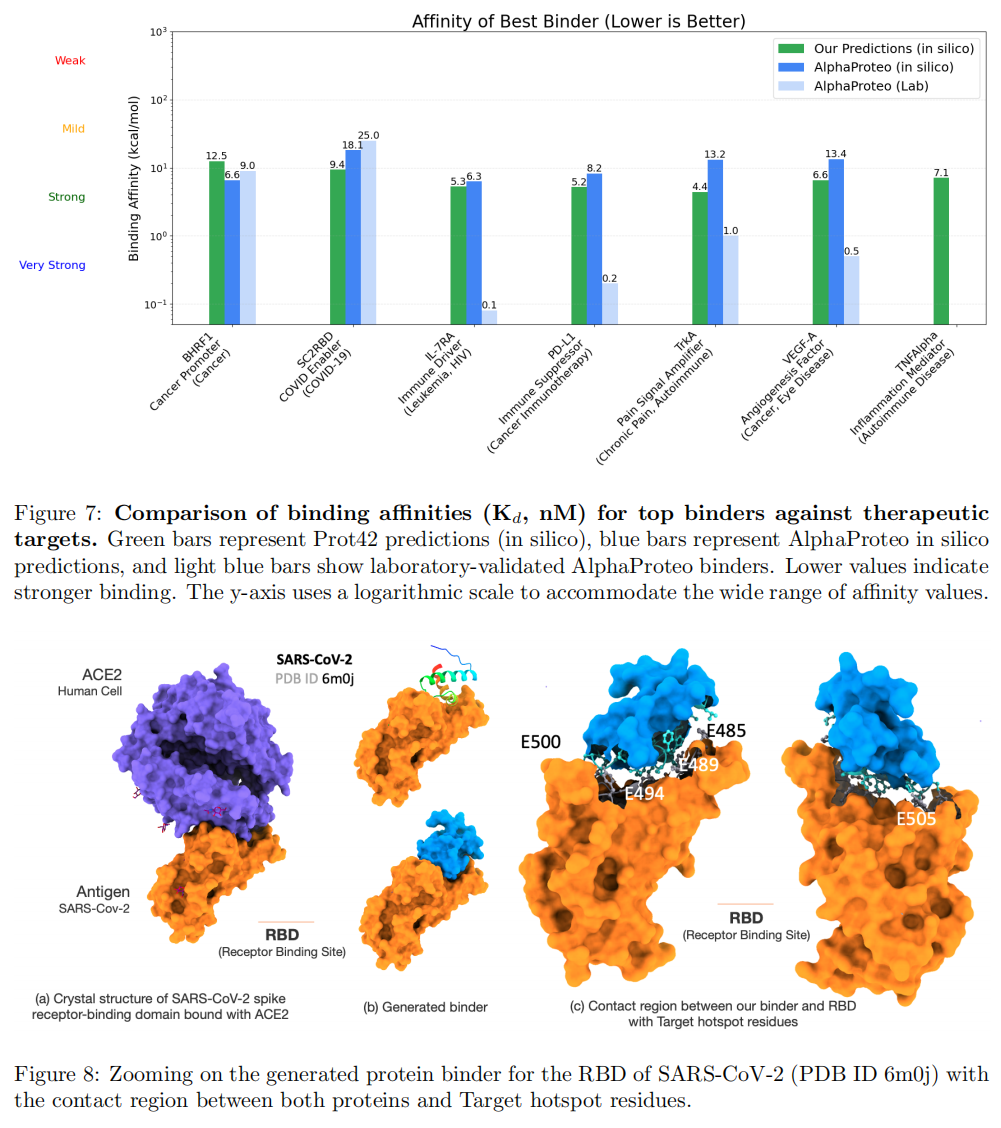
- Prot42 在所有目标上都生成了具有纳摩尔级别(nM)Kd 值的高亲和力结合体;
- 多数目标的 Kd 明显优于 AlphaProteo 的 in silico 结果;
- 尤其在 TrkA、PD-L1 和 VEGF-A 等目标上表现突出;
- 图示案例包括对 SARS-CoV-2 RBD 的结合体,能准确对接关键位点(如 E484–E505);
- Prot42 所生成结合体有望作为竞争性抑制剂,用于阻断病毒感染。
4.3 DNA Sequence-specific Binders Generation
本实验使用 PDIdb 2010 数据集中的 922 个 DNA-蛋白配对样本进行评估。
数据来源:
- 选取的结构包括 PDB ID: 1TUP、1JJ6、1A1F、8TAC 等;
- 目标是生成能识别特定 DNA 序列的蛋白质。
验证方法:
- 使用 DeepPBS 工具评估生成蛋白对目标 DNA 的结合特异性;
- 该模型使用结构、理化和几何上下文预测位置权重矩阵(PWM),从而间接衡量结合能力。
实验结果:

- Prot42 生成的结合体能够精确对接 DNA 主沟区域;
- 与参考蛋白相比,在结合位置和形状上高度一致;
- 图 9 展示了多个成功结合的三维复合体结构。

