论文地址:Scaling unlocks broader generation and deeper functional understanding of proteins
ProGen3:MOE+GLM+CLM蛋白质设计
Abstract
生成式蛋白质语言模型(Protein Language Models, PLMs)是当前用于定制蛋白质设计的强大工具,可广泛应用于医药、农业和工业等领域。尽管已有研究训练了规模不断扩大的语言模型,但关于最优训练数据分布和模型规模对生成蛋白序列的影响仍缺乏系统研究。
本文提出了ProGen3系列的稀疏生成式蛋白语言模型,并推导了计算最优的 scaling law(扩展规律),将模型扩展至460亿参数,并在1.5万亿个氨基酸 token上进行预训练。ProGen3 的预训练数据采样自精心构建的Profluent Protein Atlas v1(PPA-1),包含34 亿条全长蛋白质序列。
研究首次在湿实验(wet lab)中系统评估了模型规模对生成蛋白质序列的影响,发现更大规模的模型能够生成来自更广泛蛋白家族的可表达蛋白质。
此外,无论在计算还是实验上,结果都表明更大模型在对实验数据对齐(alignment)时表现出更高的响应性,从而在蛋白质适应度预测和序列生成能力上实现显著提升。
总体而言,这项研究表明,如 ProGen3-46B 这类大规模、基于高质量数据训练的蛋白质语言模型,可作为强有力的基础模型,推动蛋白质设计领域的研究边界。
1 Introduction
蛋白质在各类生物过程中发挥核心作用,涵盖催化反应、免疫调节、信号通路调控以及分子运输等功能,在治疗、农业、能源和工业等领域均具有重要应用价值。长期以来,功能性蛋白主要依赖于自然发现,而人工设计通常依靠随机突变与实验筛选,方法依赖性强且效率较低,难以满足特定场景下对蛋白功能的定制需求。
近年来,随着 DNA 测序成本的大幅下降,大规模天然蛋白质序列得以被采集。生成式蛋白语言模型(Protein Language Models, PLMs)因此应运而生,成为学习自然蛋白序列分布的有效工具。已有研究实验证明,PLMs 在多种实际任务中具备设计功能性蛋白的能力。
PLMs 的发展路径与自然语言处理(NLP)中的语言模型高度一致。NLP 领域已广泛验证,更大规模的模型不仅具备更强的迁移能力,还能通过精细的模型与数据扩展策略获得更优的性能。而在蛋白质建模领域,已有工作指出,PLMs 所学习的 embedding 可隐式捕捉蛋白质适应度,并能在监督或无监督场景下进行有效利用。
尽管如此,PLM 研究中仍存在若干关键问题尚未充分探索:
- 在蛋白数据量爆炸式增长的背景下,如何构建最优训练分布仍缺乏系统性研究;
- 相较于 embedding 层表征的性能评估,尚不清楚生成序列本身在模型扩展下的表现如何;
- 虽然 NLP 和蛋白建模领域均已有通过后训练策略对齐模型以满足用户偏好的尝试,但模型规模对对齐效果的具体影响尚无系统研究。
为回应上述挑战,研究者提出了 ProGen3——一组基于稀疏 Mixture-of-Experts(MoE)结构的自回归蛋白语言模型。该模型系列在保持性能的同时显著提升了训练效率。为训练 ProGen3,作者构建了规模达 34 亿全长蛋白的高质量数据集 Profluent Protein Atlas v1(PPA-1),并据此优化了训练数据分布。
在计算资源限制下,研究者推导了适用于稀疏模型的计算最优扩展规律(scaling law),并据此将模型规模扩展至 460 亿参数,在 1.5 万亿个氨基酸 token 上完成预训练。进一步地,他们首次在湿实验中系统验证了模型规模对生成蛋白质序列表达能力与多样性的影响,发现大型模型能够生成更广泛蛋白空间中的功能性序列。
最后,研究还表明,通过与实验数据对齐(如 IRPO 策略),无论模型规模如何,PLMs 均能显著提升适应度预测与序列生成能力,且模型越大,受益越显著。
2 ProGen3
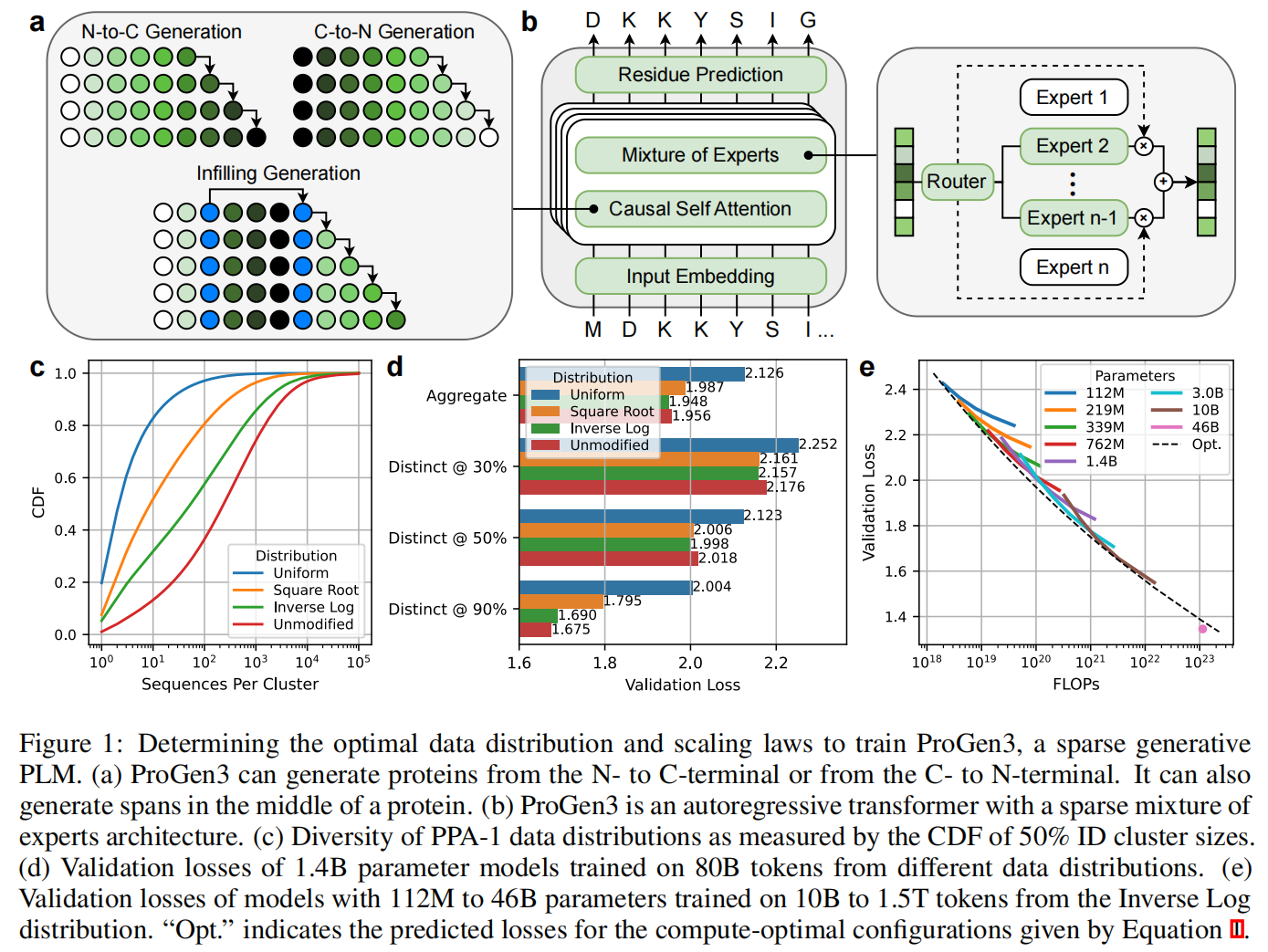
2.1 Architecture
ProGen3 是一系列自回归(autoregressive, AR)蛋白语言模型(PLMs),基于 Transformer 架构,并引入了**稀疏混合专家(sparse Mixture of Experts, MoE)**机制。每次前向传播仅激活模型总参数的约 27%(如图 1b 所示)。该模型系列涵盖从 1.12 亿到 460 亿参数的 8 种不同规模,所有模型均使用 8192 token 的上下文长度。关于架构细节,详见附录 C.1。
与 NLP 中的类似研究一致,作者发现,在固定计算预算下,稀疏模型显著优于同规模的稠密模型(详见附录 C.2)。
标准的 AR PLM 可支持因果语言建模(causal language modeling, CLM),即从 N 端(氨基)到 C 端(羧基),或反向,从 C 端到 N 端逐步生成蛋白质序列。此外,ProGen3 还支持广义语言建模(generalized language modeling, GLM),即在蛋白序列中间进行片段填充(infill)。该过程通过插入特殊哨兵 token 取代目标片段,并将这些片段及其对应哨兵标记放置在序列末尾(图 1a)。这种机制使得用户能够在保留上下文残基的情况下重新设计蛋白的局部区域。详细方法见附录 C.3。
2.2 Training Data
ProGen3 的预训练数据来自研究者自行构建的高质量蛋白质数据集 Profluent Protein Atlas v1(PPA-1),包含 34 亿条全长蛋白质序列,总计约 1.1 万亿氨基酸 token。该数据集整合了广泛的基因组和宏基因组来源,并经过多层过滤,确保其适用于训练生成式 PLM。特别地,所有蛋白片段均被排除,仅保留完整序列。整体上,PPA-1 的规模和多样性与 OMG 数据集相当,与 ESM3 的训练数据在序列数量上相近,但 ESM3 包含片段数据。PPA-1 远大于 UniRef和 BFD,更详细的数据处理流程见附录 B。
值得注意的是,尽管蛋白数据集正在迅速扩展,但其内在偏倚仍可能影响训练结果。因此,作者特别关注训练数据分布的优化问题,这是此前 PLM 研究较少涉及的方面(详见附录 A.2)。
研究者设计了四种不同的数据平衡策略,对 50% 序列相似性(ID)聚类进行采样控制,分别命名为:Uniform(均匀)、Square Root(平方根)、Inverse Log(逆对数)、Unmodified(原始分布)
这些策略控制从某个聚类(大小为 n)中采样的概率,分别与 1、$\sqrt{n}$、$\frac{n}{1 + \log n}$ 成正比。每次采样先从 50% 聚类中采样,再在该聚类内选择一个 90% ID 子聚类,最终从中均匀选取一个序列。
为了衡量模型的泛化能力,研究者构建了与训练数据无重合的验证集,分别在 30%、50%、90% ID 水平上采样,每个集合包含 250 万条蛋白质序列,均衡覆盖聚类大小(1–10, 11–100, 101–1000)范围。最终的验证损失是这三个验证集结果的平均。
如图 1d 所示,在训练 token 数固定为 80B 的情况下:
- Unmodified 分布在接近训练数据(90% ID)时表现最好;
- Inverse Log 分布在远离训练数据(30%、50% ID)时表现最佳;
- Uniform 分布效果最差,说明 PLM 仍能从频繁出现的蛋白之间学到重要信号。
基于以上分析,所有后续模型均采用 Inverse Log 分布进行训练。
2.3 Scaling Up to a 46B Parameter Model
为了在固定计算资源下最优地分配模型参数量与训练数据规模,研究者遵循 Kaplan 等人提出的扩展规律(scaling law),将验证损失建模为模型参数数量 N 与训练 token 数 D 的函数:
$$
L(N,D) = \left( AN^{-\alpha/\beta} + BD^{-1} \right)^\beta + C
$$
拟合后得出 ProGen3 的稀疏架构满足以下近似最优参数–数据对关系(图 1e):
$$
N_{\text{opt}}(D) = 2.462 \times 10^{-7} \cdot D^{1.479}
$$
基于此,计算表明:若总 FLOPs 限制为 $1.1 \times 10^{23}$,理论上应训练一个 900 亿参数模型,使用 753B token,可达到 1.376 的验证损失。但出于实际推理硬件的考虑,研究者选择训练一个 460 亿参数模型(ProGen3-46B),使用 1.5T token,预测验证损失为 1.397,实际结果更优(达到了 1.345),超过了 compute-optimal 理论边界。
他们还发现,将 warmup 训练步数从 2000 增加到 10000 可显著提升训练稳定性,这可能是实际损失优于理论预测的一个原因(附录 C.4)。
3 Computational and Experimental Analysis of Model Generations
前文已展示:随着预训练计算资源的扩展,PLMs 在验证集上的损失持续降低——无论测试蛋白是否接近训练数据。这意味着更大规模的模型更好地表示了自然蛋白分布。
然而,模型扩展对生成能力的影响此前鲜有研究。因此,本节聚焦于三种计算开销相差三个数量级的 ProGen3 模型:
- ProGen3-339M:训练于 200B tokens(1.2 × 10²⁰ FLOPs)
- ProGen3-3B:训练于 500B tokens(2.6 × 10²¹ FLOPs)
- ProGen3-46B:训练于 1.5T tokens(1.1 × 10²³ FLOPs)
3.1 Larger PLMs generate more diverse proteins that express in vitro
生成与过滤流程
作者使用 top-p 采样(p = 0.95),温度在 [0.5,1.0] 区间变化,从每个模型中无条件生成蛋白序列:ProGen3-339M:生成 4 百万条序列,ProGen3-3B:生成 3 百万条序列,ProGen3-46B:生成 2 百万条序列
为了提取“蛋白样”的序列,作者对这些生成结果施加以下质量过滤:
- 低复杂性区域过滤:移除低复杂性区域占比超过 25% 的序列(语言模型常会生成重复片段);作为参考,PPA-1 中有 95.5% 的序列通过该过滤。
- 终止 token 要求:必须包含恰当的终止符。
- 与自然序列的比对:要求序列对 PPA-1 中任一自然序列比对覆盖率大于 80%。
结果显示:
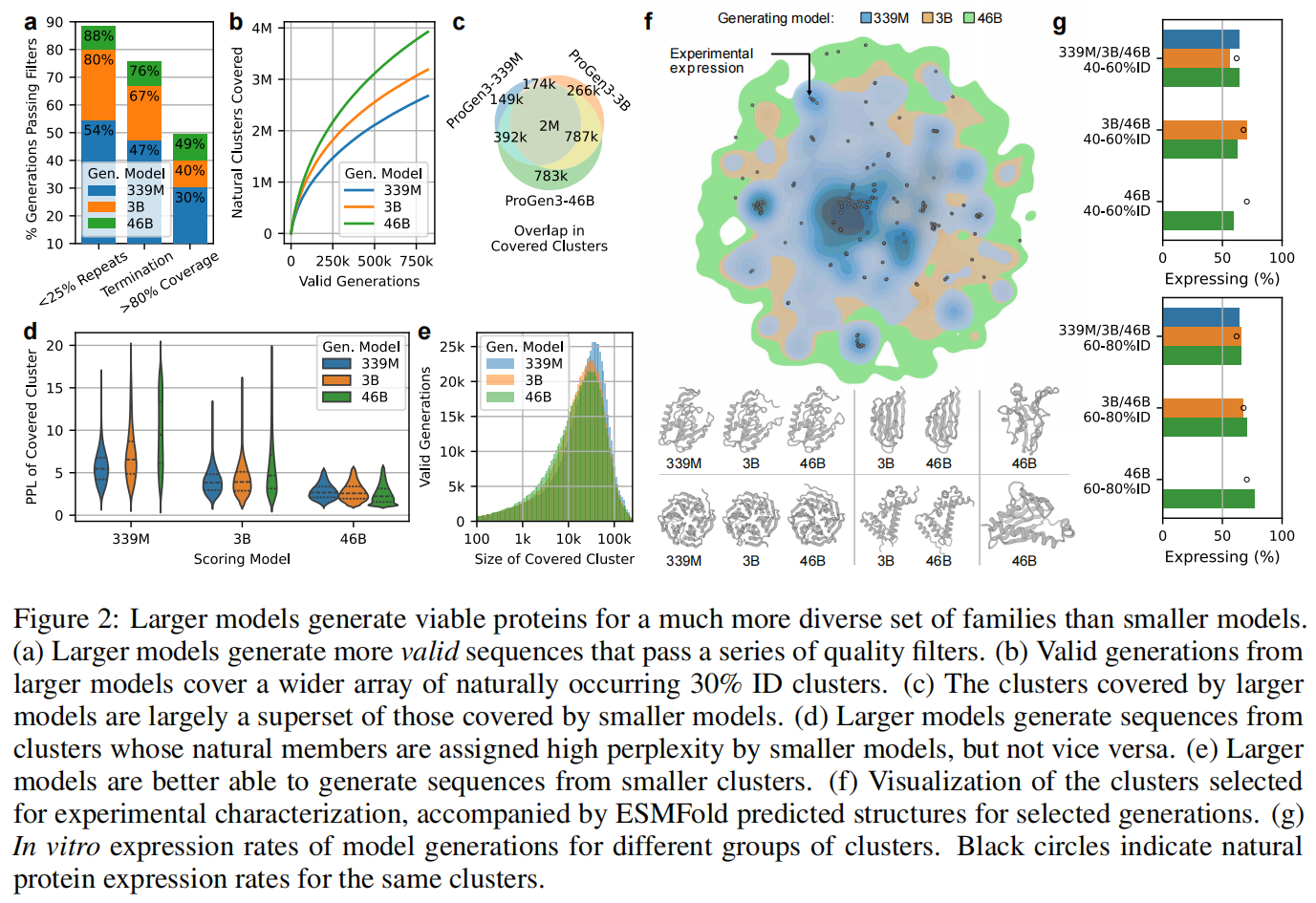
- 模型越大,通过质量过滤的序列越多;
- 从 339M 扩展到 3B 时,重复序列显著减少(见图 2a)。
结构族覆盖分析:更大模型生成更丰富的蛋白类型
接下来,作者将通过质量过滤的序列与 PPA-1 中的 30% 序列身份(ID)聚类代表比对。若某生成序列与某聚类代表的序列满足以下条件,即认为该序列“覆盖”该聚类:
- 与聚类代表的序列 ID > 30%
- 且 比对覆盖率 > 90%
⚠️ 注意:一个生成序列可能覆盖多个聚类(即便这些聚类代表间彼此 ID < 30%)。
结果:
- 更大模型生成的序列覆盖更多结构聚类(图 2b)。
- 小模型所覆盖的聚类几乎都被大模型覆盖;
- 大模型还能生成小模型完全未触及的新聚类(图 2c)。
模型生成能力是否受困于训练分布?使用困惑度解释
为回答“小模型是否能通过增加采样数量生成大模型覆盖的聚类”,作者分析了这些聚类中自然蛋白的 平均困惑度(perplexity):
- 若某模型对某个聚类中的自然序列赋予高困惑度,则说明它“不理解该聚类”,因而难以生成来自该聚类的蛋白质。
观察:
- 大模型能频繁地生成来自小模型高困惑度的聚类;
- 反之则不成立(图 2d);
- 因此,大模型可完全覆盖小模型的生成能力,但其部分生成分布对小模型而言是“分布外”的。
此外,分析显示:随着模型规模增长,其在“较小的聚类”上表现更好(图 2e),这部分解释了其拓展能力。
实验验证:体外表达评估(split-GFP 系统)
为了验证这些生成蛋白在体内是否具有“现实存在性”,作者在大肠杆菌中执行了 split-GFP 表达实验,该实验用于评估蛋白质的可溶性表达水平,其敏感指标包括:
- mRNA 表达量与稳定性、翻译效率、折叠稳定性(热力学和平衡)、抗蛋白酶能力、易聚集性、细胞毒性
split-GFP 系统与 SDS-PAGE、Western blot 等直接实验方法高度相关,且适合大规模高通量检测,已广泛用于生成模型 benchmarking
实验设计细节
从不同模型中分别选取以下类别的聚类进行实验:
- 所有三个模型都生成过的:42 个聚类
- 仅 ProGen3-3B 和 46B 生成过的:62 个聚类
- 仅 ProGen3-46B 生成过的:45 个聚类(见图 2f)
对于每个聚类:
- 每个模型各选两条序列:一条对PPA-1自然蛋白 ID 为 40–60%,一条为 60–80%
- 优先选择困惑度最低的生成序列;
- 控制组为该聚类中的两条随机自然序列;
- 所选序列长度覆盖范围为 75–300 个氨基酸,具备结构多样性。
实验结果:蛋白质表达率分析(图 2g)
- 所有模型生成序列的平均表达率相近;
- 但更大模型能在更多的聚类中生成可表达蛋白,表现出更强的泛化;
- 对所有模型而言,ID 为 60–80% 的生成蛋白表达率高于 40–60% 的;
- 有趣的是:生成蛋白与其所在聚类的自然蛋白在表达率上高度一致,说明生成序列真实可靠。
3.2 PLMs generate viable proteins outside natural sequence space
在上一节,作者已经证明:PLMs 可以在自然序列聚类中生成具备表达能力的蛋白质。
本节的目标是进一步探索:
模型是否能够在没有自然参考点的区域(即“自然分布之外”)生成功能蛋白?
作者设定了如下标准:
选取与 PPA-1 数据集中任意蛋白质的序列相似性(Identity)低于 30% 或**无法比对(unalignable)**的生成序列。
这些序列代表了模型生成能力的极端边界 —— 它们所处的序列空间中 没有自然蛋白作为“锚点”。
生成样本选取
从之前无条件生成的蛋白质中,作者按以下规则采样:
- 按照二级结构类型分为三类:
- mostly-alpha(主要为 α 螺旋)
- mostly-beta(主要为 β 折叠)
- mixed alpha/beta(混合型)
- 每种结构类别下,从每个模型中(ProGen3-339M、3B、46B)各选取 10 条序列,共 90 条样本
(见图 3a,结构预测使用的是 ESMFold)
实验验证:表达能力分析
这些 90 条序列被投入到 split-GFP 体系 中,在大肠杆菌中评估其可溶性表达水平。
作者将它们的表达结果与一个已知表达良好的“正对照蛋白”作比较(图 3b):
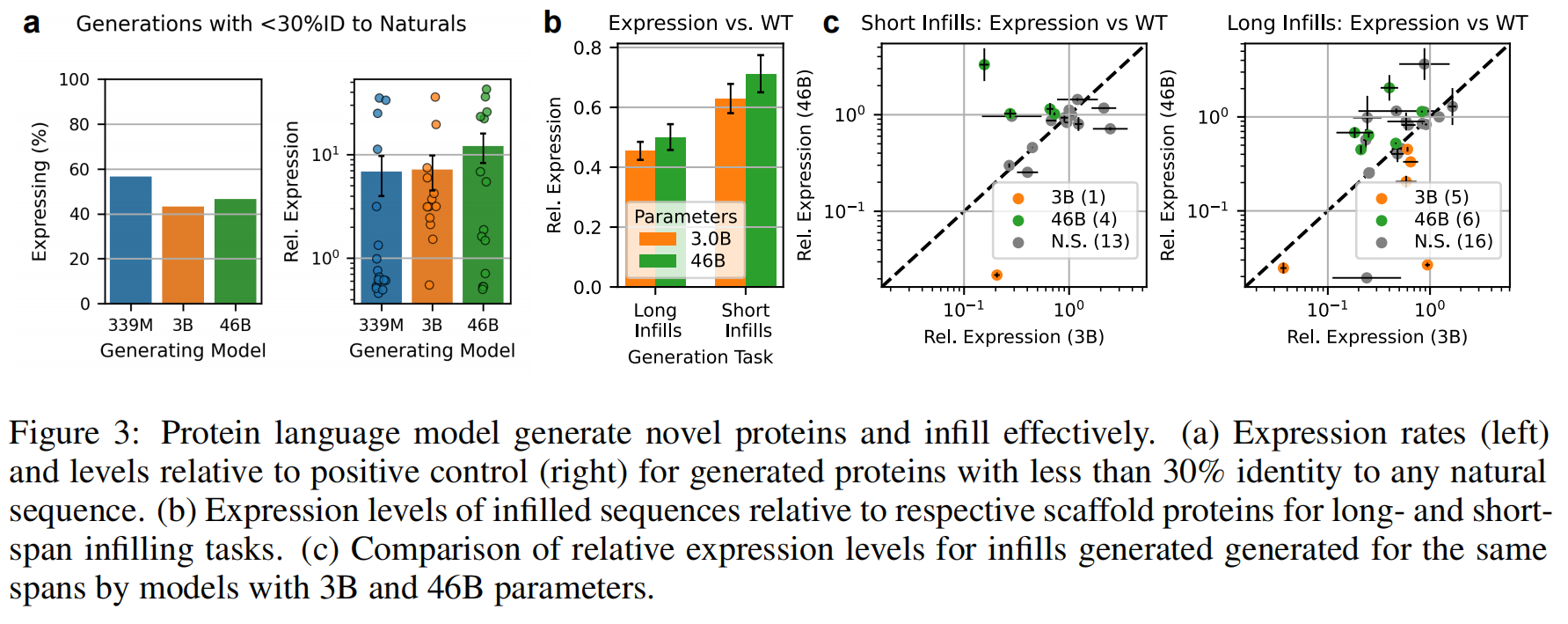
结果:
- 三个模型生成的蛋白在整体表达能力上大致相当;
- 但来自 更大模型(ProGen3-46B) 的序列在多数情况下拥有更高的表达水平,即便它们位于自然分布之外;
- 表明:大模型在生成“全新蛋白”时更有可能得到结构稳定、表达良好的结果。
3.3 Larger PLMs are better infillers
ProGen3 不仅支持传统的从头生成(causal generation),还具备强大的 “infilling”能力 —— 即在已知蛋白质序列中间的某些位置重建片段。这类能力对于蛋白设计尤为重要,例如:
- 改造活性位点、连接环(loops)、可变区等关键区域;
- 保留框架结构的同时引入功能增强突变。
为系统评估不同模型在 infilling 上的表现,作者设计了如下实验:
目标蛋白选择:
- 9 条工业/药用相关蛋白质(长度 100–400 个氨基酸):
- 这些蛋白具备一定的表达先例,来自人类或常用表达系统;
- 每条蛋白中,随机选取:2 段短片段,2 段长片段 使用 ProGen3-3B 和 46B 各自对这些片段进行 infilling。
- 9 条来自大肠杆菌胞质蛋白(cytosolic E. coli proteins)(长度 100–300 aa):
- 每条选取:1 段短片段,1 段长片段 同样由 3B 和 46B 模型分别进行 infill。
实验评估:
- 每段 in-filled 片段,从每个模型中选出 2 条序列;
- 并测量:这 2 条生成序列的表达水平;对应的天然完整蛋白的表达水平(作为 baseline);
- 使用 split-GFP 系统进行可溶性表达量检测。
表达分析与模型对比
- 在这 18 条目标蛋白中,有 11 条来源于大肠杆菌自身,理论上它们可能表现出更好的表达;
- 但这也意味着 split-GFP 对这些本源蛋白的检测存在 天花板效应,或可能偏低估计。
实验观察(图 3c):
- 总体趋势:infilling 后的表达量平均 低于天然蛋白;
- 但出现了多例 infill 片段表达 高于原始蛋白 的情况,说明 infill 并非总是“降解”功能;
- 在所有条件下,ProGen3-46B 的 infill 表达表现优于 ProGen3-3B,体现在:
- 所有片段平均表达量;
- 相同片段下 infill 的相对表达量。
4 Aligning Protein Language Models to Laboratory Data
蛋白语言模型(PLMs)通常基于进化数据进行预训练,为评估其泛化能力,研究者常采用 零样本适应度预测(zero-shot fitness prediction) 的标准任务:
- 任务形式:输入突变后的蛋白序列,让模型基于其概率/似然性进行评估;
- 评估方式:将模型对不同突变序列给出的 似然(log-likelihood) 与实验中测得的 适应度评分 做 Spearman 等级相关系数(ρ) 分析;
- 数据来源:通常使用深度突变扫描(Deep Mutational Scanning, DMS)数据集,如 ProteinGym
问题:大模型验证损失虽低,但零样本预测能力下降
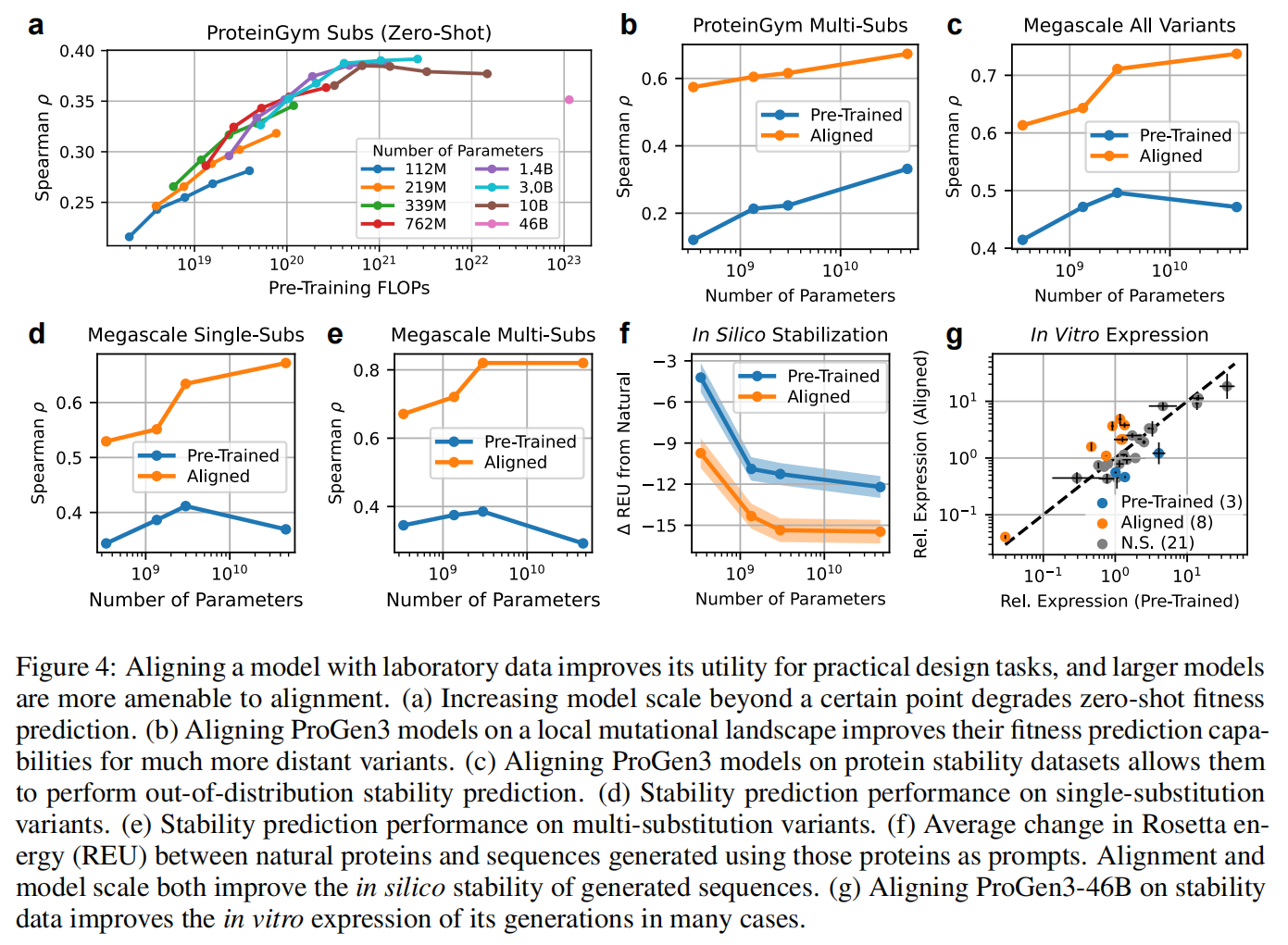
作者发现了一个有趣现象:
尽管更大规模的 ProGen3 模型(>3B 参数)在验证损失和序列多样性上表现更优,
但它们在 ProteinGym 的零样本预测中表现反而变差(图 4a)。
这一发现印证了已有文献中的两个重要假设:
- Weinstein et al.和 Gordon et al.曾指出:
- 在某个临界点之后,越能精确刻画自然分布的模型,反而越不利于预测突变的功能效应;
- 原因在于:自然蛋白演化过程中的协变量结构可能与突变适应度不一致,导致预训练目标(建模分布)与评估目标(功能预测)出现张力。
应对策略:通过对齐显式引导模型能力
尽管大型模型在零样本设置下表现不佳,但作者认为它们具备可唤醒的潜在能力(latent ability),只需合适地引导即可显现。
为此,作者引入了 IRPO(Iterative Reasoning Preference Optimization):
- IRPO 是一种受大语言模型对齐启发的策略,旨在将模型的原始 log-likelihood 分布进行微调,使其更贴合实验测得的目标属性。
- 在本研究中,IRPO 用于对齐的实验指标包括:酶活性(activity),分子结合能力(binding),有机体适应度(organismal fitness),蛋白稳定性(stability)
4.1 Supervised Fitness Prediction
作者希望展示:
经过 IRPO alignment 的 PLM 能在仅使用本地突变(局部训练)信息的情况下,实现对远距离突变蛋白的准确预测,从而提升定向进化(directed evolution)的效率。
实验设置(ProteinGym)
- 使用 ProteinGym 数据集,其中包含多个深度突变扫描(DMS)实验;
- 选择包含 ≥3 位点突变的所有实验(assays);
- 训练数据:包含与野生型相差不超过 k 位突变的所有变体;k 是使得训练集 数量超过 500 条序列的最小值;
- 为确保训练/测试集的适应度分布相似,设置约束:训练与测试集中适应度分布的总变差(Total Variation Distance) < 1。
最终筛选得到 8 个测量不同蛋白功能属性的 assay,涵盖广泛蛋白种类(见补充表 8)。
结果一:训练局部,预测远程,ProGen3 泛化能力显著
- 使用不到 500 条单突变体的实验数据,IRPO 对齐后的模型就能准确排序相距十数个突变的远程变体;
- 模型规模越大,预测能力越强(图 4b,补充图 6):
- ProGen3-46B(ρ = 0.673) 优于 KERMUT(ρ = 0.628)
- 与 SOTA 方法 ConFit(ρ = 0.679) 相当;
- 相比 KERMUT 和 ConFit 等专为预测突变效应设计的模型,ProGen3 作为一个通用的生成模型,不依赖结构信息或特定手工特征,仍能达到竞争性能;
- 值得注意的是,IRPO 可用于优化替换突变、插入和缺失的适应度评估,而非仅限于 substitution
实验扩展:预测蛋白稳定性(ΔG)
- 将 IRPO 应用于 Megascale 蛋白稳定性数据集:
- 包含近 80 万个单/双突变体,分布于 479 个蛋白结构域;
- 每个样本均标注了折叠自由能(ΔG);
- 使用 FoldSeek easy-cluster(比对覆盖率 ≥50%)对蛋白结构域进行聚类:留出 5% 聚类用于验证,5% 用于测试;
- 模型仅在单点突变体上进行训练,以检验其泛化能力。
结果二:大模型可在结构未知场景中精准预测蛋白稳定性
- 对齐后的 ProGen3 模型在蛋白稳定性预测任务上同样随着规模提升而表现更好(图 4c–e);
- 与结构感知模型 ProteinDPO对比:
- 在相同训练/测试划分下:
- ProteinDPO:ρ = 0.72
- ProGen3-46B(IRPO):ρ = 0.737
- 尽管 ProGen3 是基于序列的模型,在未见的双突变体上具有更强的泛化能力:
- 多突变体预测上:ProGen3-46B:ρ = 0.820
- 同条件下:ProteinDPO 仅为:ρ = 0.468(图 4e)
- 在相同训练/测试划分下:
4.2 Sequence Generation
在前文验证了对齐策略(IRPO)显著提升了 ProGen3 的适应度与稳定性预测能力后,作者进一步评估: 对齐后的模型在蛋白生成任务中的表现是否也更优?
序列生成设置
作者从第 3.1 节中使用的自然蛋白中,挑选出 32 条结构多样的自然蛋白质作为生成提示(prompts),这些蛋白长度在 98–282 个氨基酸之间,远长于 Megascale 稳定性数据集中 40–72 氨基酸的蛋白结构域。
对于每个 prompt,他们使用 稳定性对齐模型与原始预训练模型分别进行生成:
- 每种模型、每个 prompt,各生成 50,000 条序列;
- 每条序列需通过图 2a 中定义的质量过滤标准;
- 使用两种条件化方式生成:
- 用前 25% 条件化生成后 75%;
- 用后 25% 条件化生成前 75%;
- 最终,从每组(模型 × prompt)中选出 100 条困惑度最低且通过质量过滤的序列,进入后续分析。
结构与稳定性评估(计算模拟)
作为初步评估,作者使用 in silico 稳定性预测流程:
- 对所有生成序列及其对应的自然 prompt 序列使用 ESMFold 预测三维结构;
- 在 PyRosetta 中进行:4 轮最小化(MinMover);4 轮结构松弛(FastRelax);每个序列进行 5 条独立轨迹;
- 记录每个序列在所有轨迹中达到的最低 Rosetta 能量值;
- 与对应自然序列的最低能量值做差值计算,差值越低表示生成序列更稳定。
结果显示(图 4f):
- 更大模型生成的蛋白质平均更稳定;
- 经过 IRPO 对齐后,所有模型的稳定性进一步提升;
- 说明 alignment 不仅优化了模型预测能力,也显著改善了其生成能力。
湿实验验证(split-GFP 表达实验)
前人研究已表明,split-GFP 表达水平与蛋白稳定性显著正相关。
为进一步验证 alignment 是否确实提高了生成序列的稳定性,作者设计以下实验流程:
- 对每个 prompt,从两个模型(原始 ProGen3-46B 和对齐版)中各选择:2 条困惑度最低的生成序列;
- 使用 split-GFP 实验系统测量蛋白质表达水平;
- 表达结果以对应的野生型蛋白为基准归一化,进行比较(图 4g);
- 使用 配对样本 Welch’s t 检验(p < 0.01)评估差异显著性。
实验结果:
- 在 32 个 prompt 中:alignment 提高表达的有 8 个;降低表达的有 3 个;无显著影响的有 21 个;
- 显示 alignment 对于一部分蛋白序列的生成质量有显著提升,且在多数情况下并不会带来负面影响。

