论文地址:Predicting a Protein’s Stability under a Million Mutations
MutateEverthing:大模型借突变点位MLP预测高点
Abstract
稳定蛋白质是蛋白质工程的基础步骤 。然而,由于现有蛋白质的进化压力,识别少量能改善热力学稳定性的突变非常具有挑战性 。深度学习最近已成为识别有前景突变的强大工具 。但现有方法计算成本高昂,因为模型推理的次数与查询的突变数量成正比 。
该研究的主要贡献是一种简单、并行的解码算法,名为“Mutate Everything” 。该算法能够在一次前向传播中预测所有单点和双点突变的影响 ,甚至能以极小的计算开销预测更高阶的突变 。研究人员将“Mutate Everything”构建在ESM2和AlphaFold模型之上,这两个模型原本均未针对预测热力学稳定性进行训练 。他们在Mega-Scale cDNA蛋白水解数据集上进行了训练,并在S669、ProTherm和ProteinGym数据集的单点和高阶突变预测中取得了当前最佳的性能 。
1 Introduction
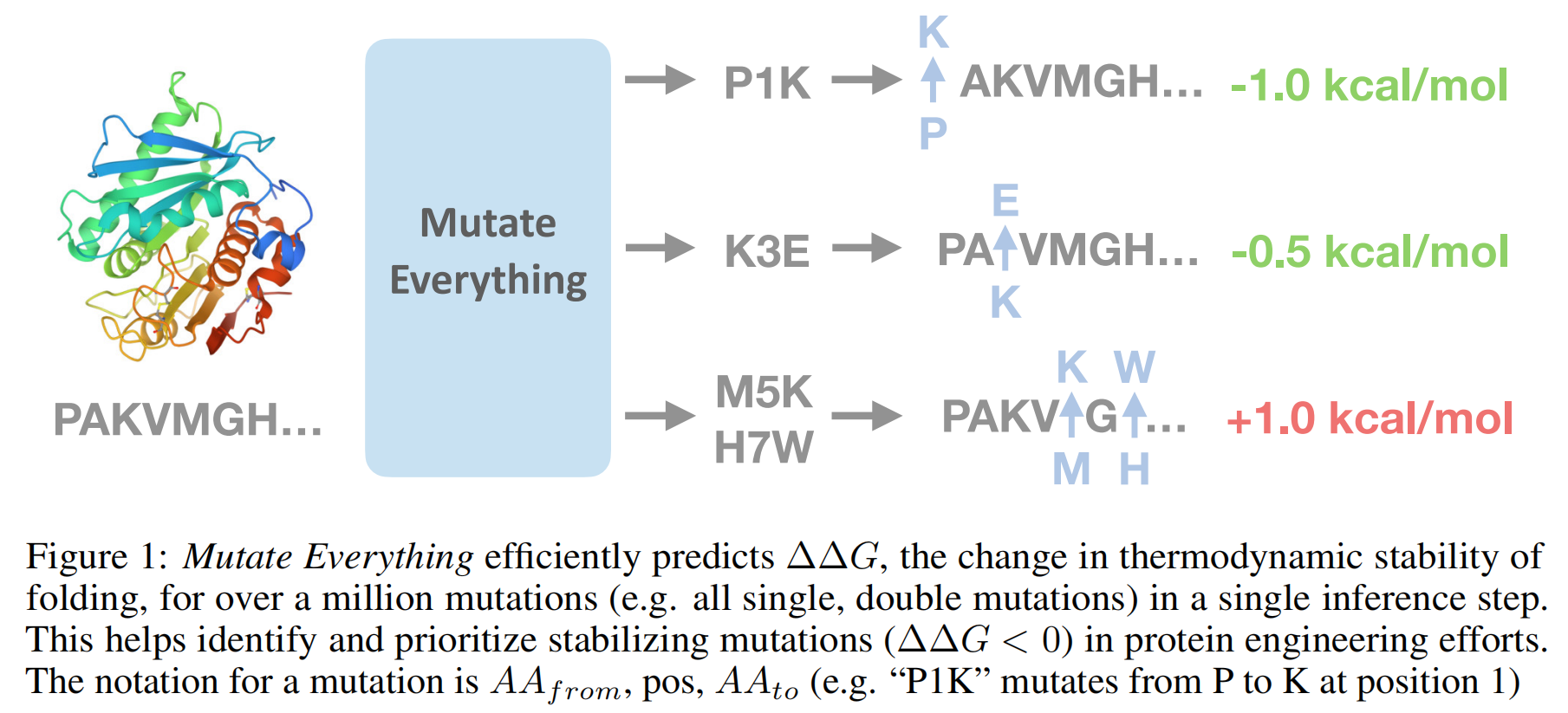
蛋白质工程是一门通过改变天然蛋白质序列以改进其特性,从而服务于工业 和制药应用 的学科。 然而,进化过程同时优化了蛋白质在其原生环境中的多种特性,这导致了蛋白质通常只具有临界的热力学稳定性(大约在5到15千卡/摩尔的范围内)。这种临界稳定性使得这些天然蛋白质在被置于要求更高的工业环境中时,往往会变得没有功能 。因此,当需要将一个天然蛋白质重新用于生物技术应用时,工作流程通常必须从识别那些不会对蛋白质造成损害(即非有害的)并且能够稳定其结构的突变开始 。一旦蛋白质拥有了一个经过稳定化的结构,那么后续的、下游的工程目标——这些目标往往需要进一步探索那些可能会降低稳定性的突变——才变得切实可行和易于管理 。
从历史上看,这个稳定化蛋白质的过程一直受到一些瓶颈的制约。这些瓶颈主要来自于对理性设计的候选突变体进行广泛且耗时的实验室表征的需求 ,或者是来自于构建和筛选指导性进化库的复杂过程 。
近年来,随着高通量实验技术的发展,生物数据领域出现了爆炸式的增长 。这种数据的极大丰富使得深度学习框架得以被应用于加速识别那些具有稳定化效果的突变的过程 。一个成功稳定化的蛋白质往往并不仅仅依赖于单一的突变,而是通常需要引入多个突变才能达到理想的稳定性水平 。然而,目前存在的深度学习框架在预测突变效果时,大多没有充分考虑到多个突变同时存在时可能发生的上位性相互作用(epistatic interactions)。因此,为了能够显著地加速蛋白质工程的整体进程,一个至关重要的方面是能够高效地探索和理解高阶突变(即包含多个同时发生的点突变的组合)所形成的复杂上位性图谱 。但是,由于高阶突变具有内在的组合特性——即可能的突变组合数量会随着突变位点数的增加而急剧上升——因此,对其进行彻底的、详尽的探索很快就会在计算上变得不切实际且成本过高 。
先前预测热力学稳定性的模型通常会为输入序列生成嵌入或表示 。该论文提出的解码器则基于这些表示,通过一个线性网络为每种可能的单一突变创建进一步的表示 。这些突变级别的表示随后被聚合以形成高阶突变的表示,并输入一个轻量级的多层感知器(MLP)头部以预测 ΔΔG 值 。由于突变级别的表示仅计算一次,该模型能够高效扩展至数百万级别的(高阶)突变,因为聚合和MLP头部的计算成本低廉 。因此,在固定的计算预算下,该模型能比先前方法评估多出数百万个潜在突变 。
Mutate Everything 能够在单个GPU上于数秒内估算出单个蛋白质所有单一和双重突变的影响,从而有潜力高效计算整个人类蛋白质组中所有约20,000个蛋白质的单一和双重突变体的稳定性 。据论文作者所知,Mutate Everything 是首个使整个蛋白质组双重突变体计算分析变得易于处理的工具 。该方法可与任何能生成蛋白质序列表示的模型结合使用 。论文中,作者采用 AlphaFold 作为骨干架构,并在一个通过实验衍生的 ΔΔG 值标记的数据集上对其进行微调,从而产生了首个基于 AlphaFold 的精确稳定性预测模型 。
研究者们在多个成熟的基准数据集(如 ProTherm、S669 和 ProteinGym)上评估了 Mutate Everything 的性能 。在ProTherm的高阶突变子集(PTMul)上,该模型实现了0.53的Spearman相关系数,优于次优方法的0.50 。在S669的单一突变上,模型达到了0.56的Spearman相关系数,超越了先前0.53的最佳水平 。在ProteinGym上,Mutate Everything将现有最佳方法的Spearman相关系数从0.48提升至0.49 。该模型使寻找最稳定的双重突变在计算上变得可行,并在cDNA蛋白水解数据集的双突变子集(cDNA2)上证明了其能有效将稳定突变优先排序的能力,即便在该数据集中仅有3%的已知突变为稳定突变 。
3 Preliminary
Problem Setup
该研究的核心目标是针对大量的给定突变集 (M1,…,MN),确定蛋白质 w 在引入这些突变后其热力学稳定性发生的变化,这个变化用实数 ΔΔG∈R 来表示 。这里的每一个突变集可以包含单个突变也可以包含多突变,后者即所谓的高阶突变 。
此外,现有的方法范式在需要高效扩展到处理大量突变(例如数百万个)时,往往显得力不从心 。 那些能够处理高阶突变(即包含多个突变)的模型,通常的工作方式是接收一个蛋白质及其对应的突变体序列作为输入,然后预测两者之间的稳定性差异 。 采用这类方法时,如果想要从一个相对较小的蛋白质(例如,一个包含75个氨基酸的蛋白质,其所有单点和双点突变可能超过一百万个)中找到最能提高稳定性的突变,就需要进行超过一百万次的独立模型评估和后续的排序工作,这在计算上是非常耗时的 。本论文提出的模型核心思想是仅需对一个经过微调的骨干网络(backbone)进行一次前向传播计算,然后通过一个轻量级的解码器(decoder)对所有突变并行地进行一次解码操作 。
Feature Extraction
在特征提取方面,为了缓解实验标记数据相对稀缺的问题,研究者们采用了微调(fine-tune)AlphaFold以及其他预训练模型的方法 。 AlphaFold模型中的Evoformer模块能够学习蛋白质序列中的共进化模式,例如不同残基之间的相互作用信息;而其结构模块(Structure Module)则能学习与蛋白质三维结构相关的信息,比如残基的溶剂可及性或残基之间的距离等 。 论文的研究表明,这些从AlphaFold中提取的特征与蛋白质的稳定性以及突变如何影响这种稳定性之间存在紧密的联系 。
4 Methodology
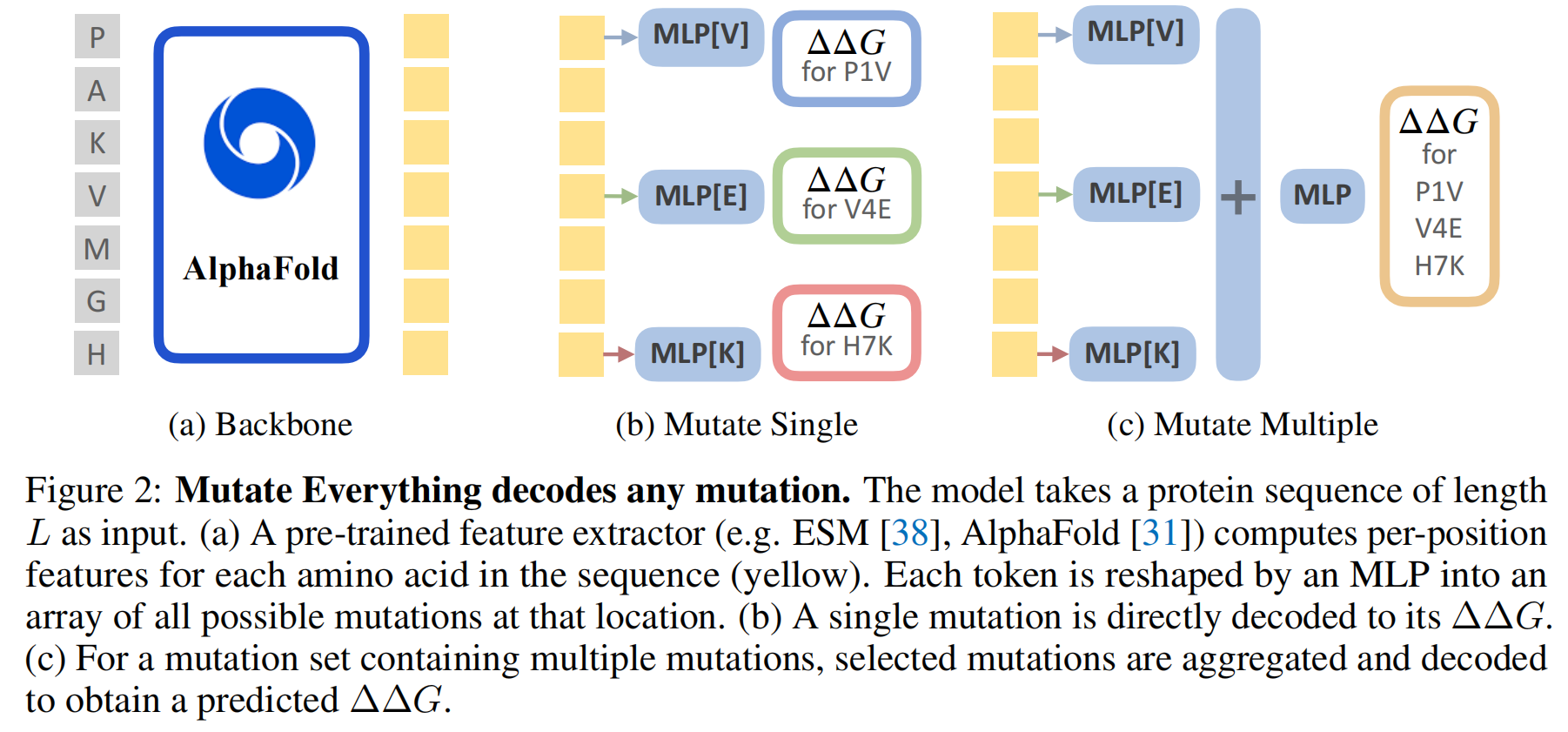
对于每一个在位置 p 变为氨基酸 t 的突变 $μ=(p,t)$,模型会计算一个潜在的突变表示 $z(μ)∈R^d$,该表示旨在捕捉该突变对蛋白质的影响 。这个突变表示被分解为 $z(μ)=f^t(x_p)+h^t$ 。其中,依赖于序列的特征 $f^t∈{f^A,…,f^Y}$ 会根据突变后的氨基酸类型来投射该位置的特征 $x_p$ 。而独立于序列的特征 $h^t∈{h^A,…h^Y}$ 则是对应不同氨基酸类型(A, C,…, Y)的独有嵌入 。直观地理解,$f^t$ 捕捉了该位置的上下文相关特征,而 $h^t$ 则捕捉了关于突变后氨基酸类型的信息 。这种突变表示构建了一个代表氨基酸替换的特征 。
Decoding single mutations
“解码”在这里的意思是将前面得到的数学化突变表示 z(μ) 转换成一个具体的、可理解的预测值,也就是蛋白质热力学稳定性的变化量 ΔΔG 。
这个转换过程由一个轻量级的线性头部(lightweight linear head)g1 完成 。这个“头部”本质上是一个小型的神经网络层,它学习如何从 z(μ) 映射到 ΔΔG 值 。
Decoding higher-order mutations
图2c阐释了高阶突变的解码过程 。求和一个高阶突变中每个单点突变的表示
聚合之后,同样会有一个轻量级的解码头部 g (与单点突变解码器结构类似但可能参数不同) 来处理这个聚合后的表示,从而得到高阶突变的预测稳定性变
对于高阶突变(K>1),研究者们通过实验发现了一个有益的技巧:模型预测的值并非直接是最终的 ΔΔG,而是相对于“构成该高阶突变的各个单点突变的 ΔΔG 预测值之和”的那个残差(residual)。也就是说,模型学习的是实际多点突变效应与简单加和的单点突变效应之间的差异,这更能捕捉到非线性的上位性效应。具体效果可以参见论文中的表5d 。
Decoding a million mutations in one forward pass
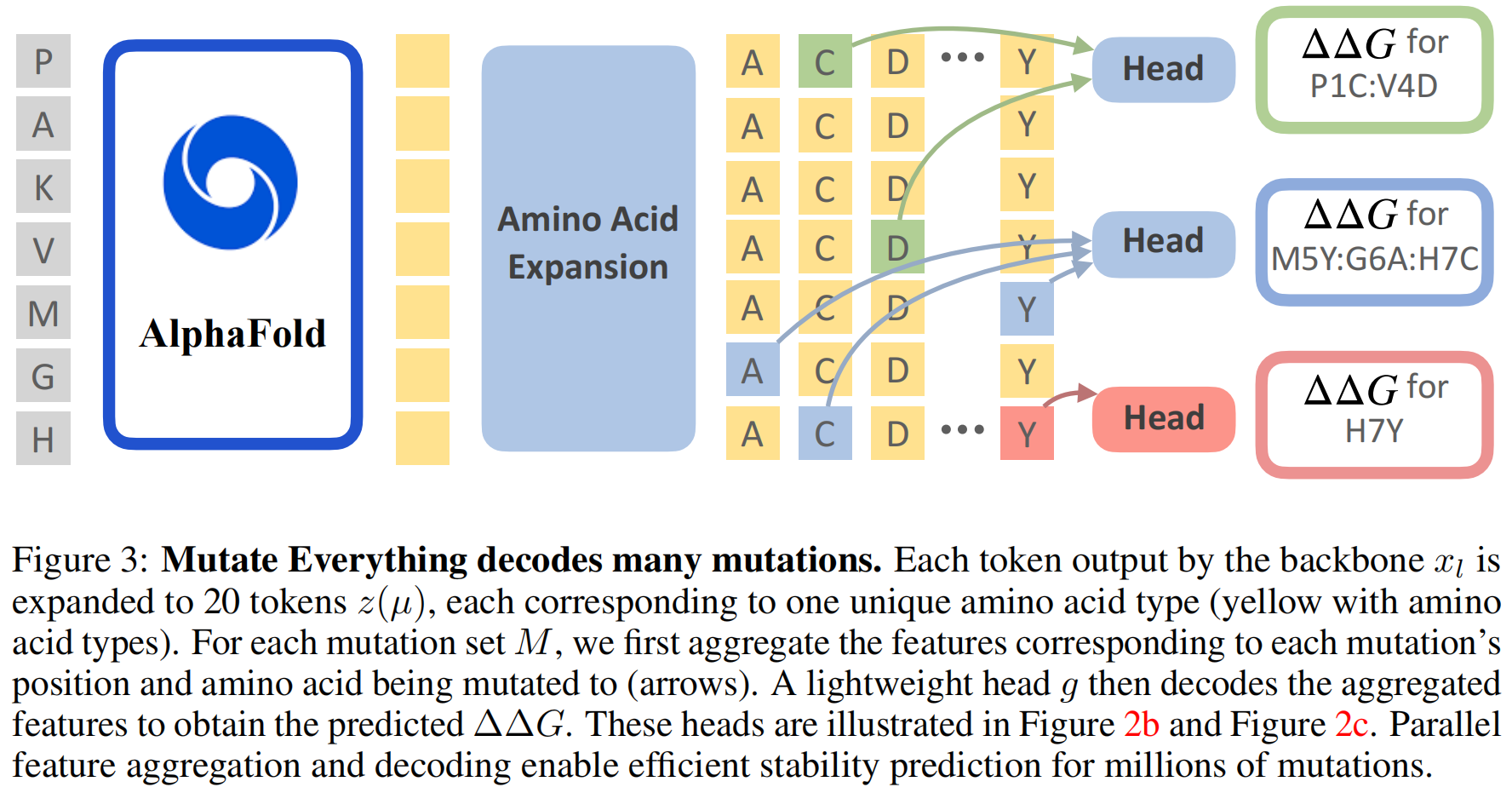
这部分是该方法高效性的核心所在,能够同时高效计算数百万个突变的 ΔΔG 值(如图3所示)。其关键思想在于:对一个蛋白质,预先计算出所有单个位置变成所有20种氨基酸的突变特征,然后在后续的预测中重复利用这些预计算好的特征
具体步骤如下:
- 首先,对于一个长度为 L 的蛋白质序列,模型会计算出所有 L×20 种可能的单点突变表示,形成一个集合 Z={z((p,t)):p∈[1,…,L],t∈AA}。这里 AA 代表20种氨基酸。这意味着,原始序列中每个位置的特征 xl 都会被扩展成20个不同的突变表示,分别对应它突变成20种氨基酸中每一种的可能性(包括不突变,即变成它自己——野生型氨基酸)。
- 对于一个特定的蛋白质序列,上述的骨干网络特征提取和这种突变表示的扩展过程只需要执行一次。
- 然后,当需要预测任何一个突变集(无论是单点还是高阶)的 ΔΔG 时,模型会从预先计算好的 Z 中索引(查找)出这个突变集里包含的那些特定单点突变的表示,并将它们加和起来 。这些索引和求和操作因为是基于预计算结果,所以非常快速,并且可以被高度并行化处理,即同时为许多不同的突变集进行计算 。论文提到,当 K=2(双点突变)时,使用外和(outer-summation)操作实现并行化;当 K>2 时,使用“收集”(gather)操作 。
- 最后,那个轻量级的解码器 g 会并行地为每一个(聚合后的)突变集表示计算出其对应的 ΔΔG 值 。
Training
优化的目标函数是Huber损失(Huber loss)。Huber损失是一种在均方误差(MSE)和平均绝对误差(MAE)之间的折中,它对数据中的异常值(outliers)不如MSE那么敏感
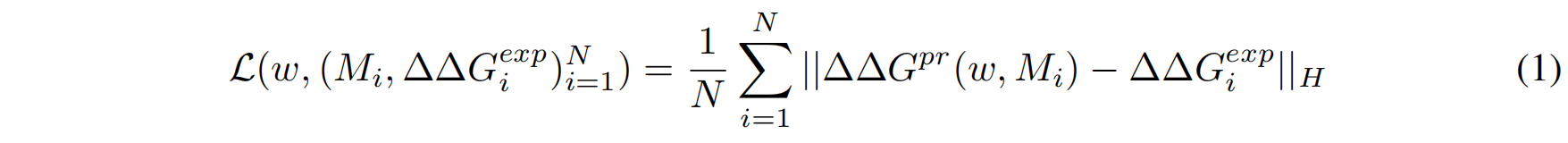
这种训练方式的一个重要优点是,对于一个给定的蛋白质,其所有已标记突变的损失可以通过一次反向传播(backpropagation)过程进行高效计算和更新,这与以往那些可能需要对每个突变单独进行一次反向传播的工作相比,效率更高 。模型训练时使用的数据是单点和双点突变体,平均每个蛋白质大约有2000个突变数据点 。
训练数据中存在一个现象:只有大约3%的双点突变是稳定化的(即 ΔΔG<−0.5 kcal/mol)。为了防止模型因为绝大多数是不稳定或中性突变而对这些少数但非常重要的稳定化突变学习不足,研究者们对训练数据进行了处理:他们对那些非稳定化的双点突变进行了子采样(subsample),使得它们的数量最多是被标记为稳定化的双点突变数量的8倍 。这样做有助于模型更好地学习和识别那些稀有的稳定化突变
5 Results
5.1 Implementation Details
主要的特征提取器是 AlphaFold,具体实现基于 OpenFold 。所需的多序列比对(MSA)是使用 Colabfold 生成的 。来自 AlphaFold 的 Evoformer 和结构模块(Structure Module)的序列特征被聚合起来作为解码器的输入 。模型中还使用了一个适配器(adapter)将骨干网络的隐藏层维度映射到 D=128 。氨基酸投射和单点突变解码器是通过线性层实现的 ,而高阶突变解码器则使用一个2层MLP转换先前的嵌入 ,这些表示被聚合后送入一个3层MLP来预测 ΔΔG 。
模型的训练数据来源于 cDNA display proteolysis dataset 。这是一个利用高通量蛋白水解筛选结合下一代测序(NGS)技术获得的数据集,包含了100个微小蛋白质(mini-proteins)的超过10万个单点和双点突变的ΔΔG的带噪测量值 。为了评估模型对未见过蛋白质的泛化能力,研究者从训练集中移除了与评估基准中任何蛋白质具有高序列相似性的蛋白质(使用MMSeqs2进行比较) 。
训练过程包括:首先在单点突变上微调预训练的骨干网络20个epoch 。然后,再在单点和双点突变上共同微调模型100个周期,使用带有10个预热周期的余弦学习率调度策略(cosine learning rate schedule) 。由于AlphaFold的高内存需求,批处理大小(batch size)设置为3个蛋白质 。学习率设置为 3×10−4,权重衰减(weight decay)为0.5 。整个训练过程在3个A100 GPU上耗时约6小时 。
5.2 Finding Stabilizing Double Mutations
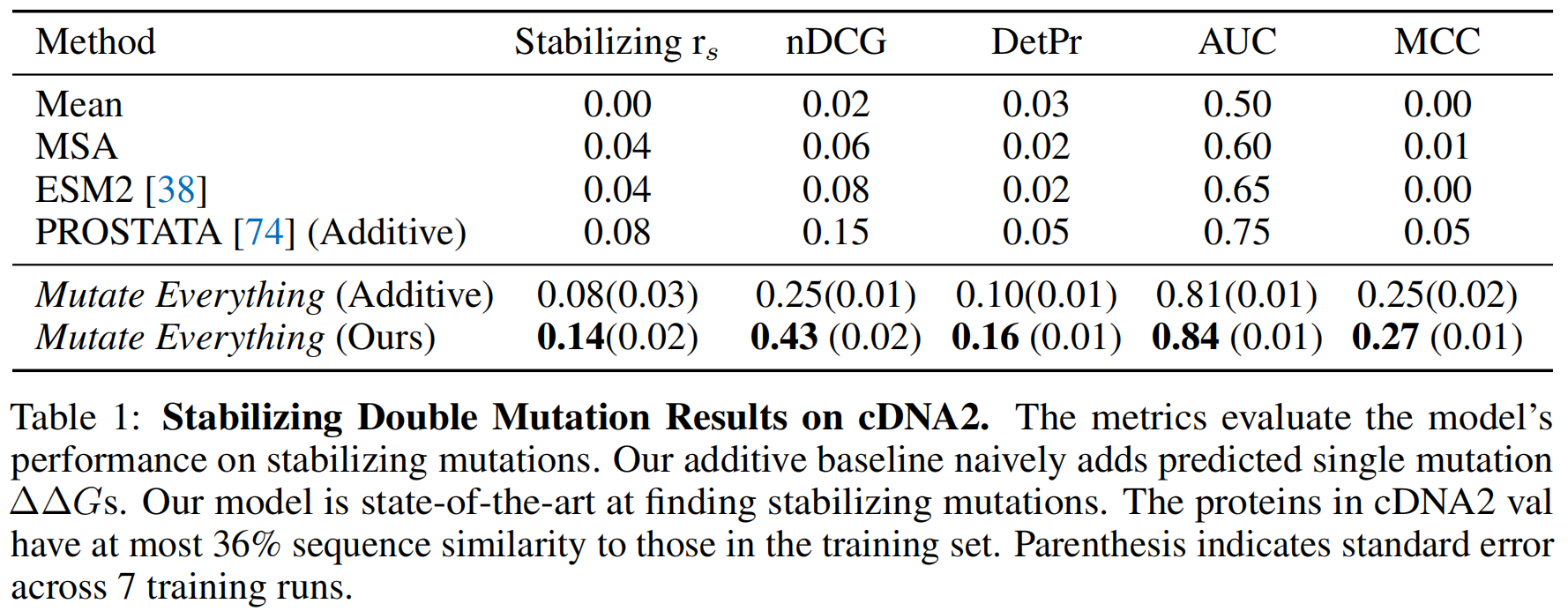
这部分评估了模型在cDNA2数据集上寻找稳定化突变的能力 。cDNA2是cDNA双突变数据集的验证分割,包含18个微小蛋白质,总计22,000个双突变 。该验证集中的蛋白质与cDNA训练集中的蛋白质同源性最高不超过36% 。在这些突变中,只有198个(约0.8%)是稳定化的(ΔΔG<−0.5 kcal/mol) 。
表1展示了不同方法在稳定化突变上的评估结果 。归一化折损累积增益(nDCG) 通过考虑实验验证的ΔΔG值及其在排序列表中的位置来衡量排序突变列表的质量 。检测精度(DetPr) 是指在模型预测的前 K=30 个突变中,实验验证为稳定化突变的比例 。
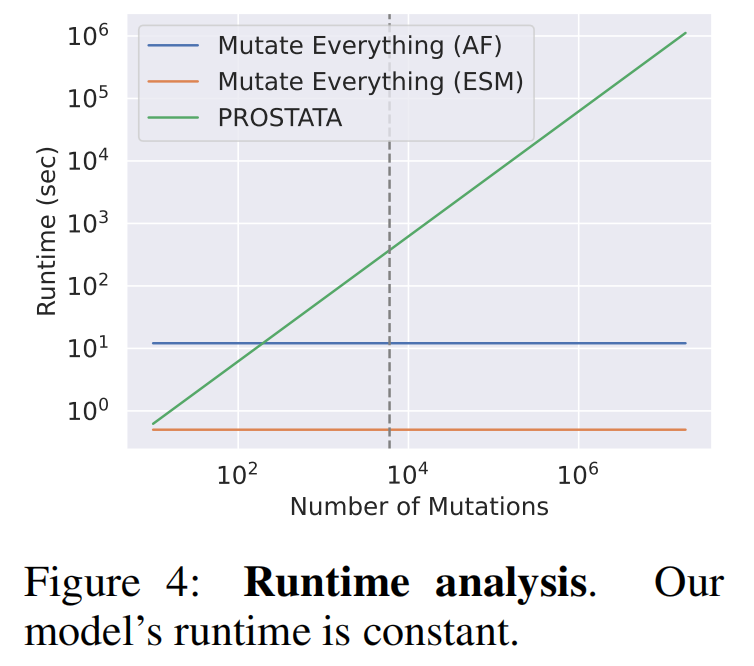
图4展示了几种方法在一个包含317个氨基酸的蛋白质上,于A100 GPU上的运行时间 。图中的虚线表示从评估单突变到双突变的转换点 。Mutate Everything在模型的单次前向传播中即可预测所有单点和双点突变的ΔΔG 。使用ESM2作为骨干网络时,运行时间为0.6秒;使用AlphaFold作为骨干网络时,为12.1秒 。相比之下,同样使用ESM2骨干网络的PROSTATA方法,由于需要将蛋白质序列及其突变序列作为输入来输出稳定性变化,评估所有双突变需要306小时(批处理大小为768),即使在8个GPU的节点上也需要1.5天 。而Mutate Everything的运行时间是恒定的,不受突变数量的影响
5.3 Higher-order Mutation Results
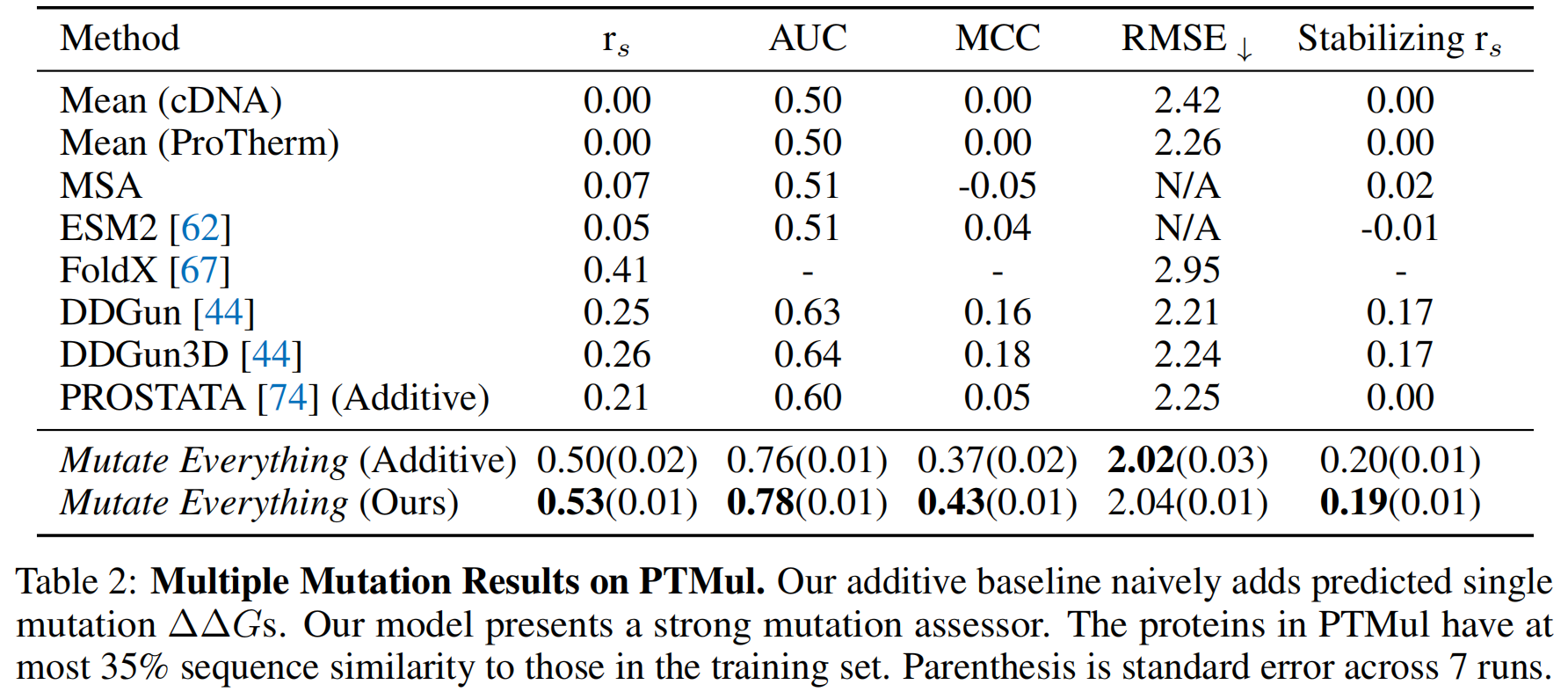
这部分评估了模型在ProTherm数据集中预测高阶突变(即包含多个氨基酸变化的突变)引起的热力学稳定性变化的能力 。ProTherm是一个收集了文献中蛋白质突变的实验热力学数据(ΔΔG)和热稳定性数据(ΔTm)的数据库 。研究中使用了该数据库的一个子集,名为ProTherm Multiple (PTMul),其中包含高阶突变的ΔΔG值,共有858个突变 。在移除了4个重复和8个模糊突变后,保留了846个实验验证的高阶突变ΔΔG值,其中包括536个双突变和183个三点突变 。PTMul中的蛋白质与训练集中使用的蛋白质同源性最高不超过35%
表2展示了在PTMul上的结果 。其中,加性基线(additive baselines)是通过简单地将单个突变的预测ΔΔG值相加得到的 。这个基线严格地评估了模型学习上位性(epistasis,即组合突变时非加性的相互作用)的能力 。结果显示,Mutate Everything在ProTherm上取得了0.53的Spearman相关系数rs的SOTA性能,而其加性基线的表现为0.50 。表6中还提供了PTMul在双突变和更多(>2)突变上的细分性能
5.4 Single Mutation Results
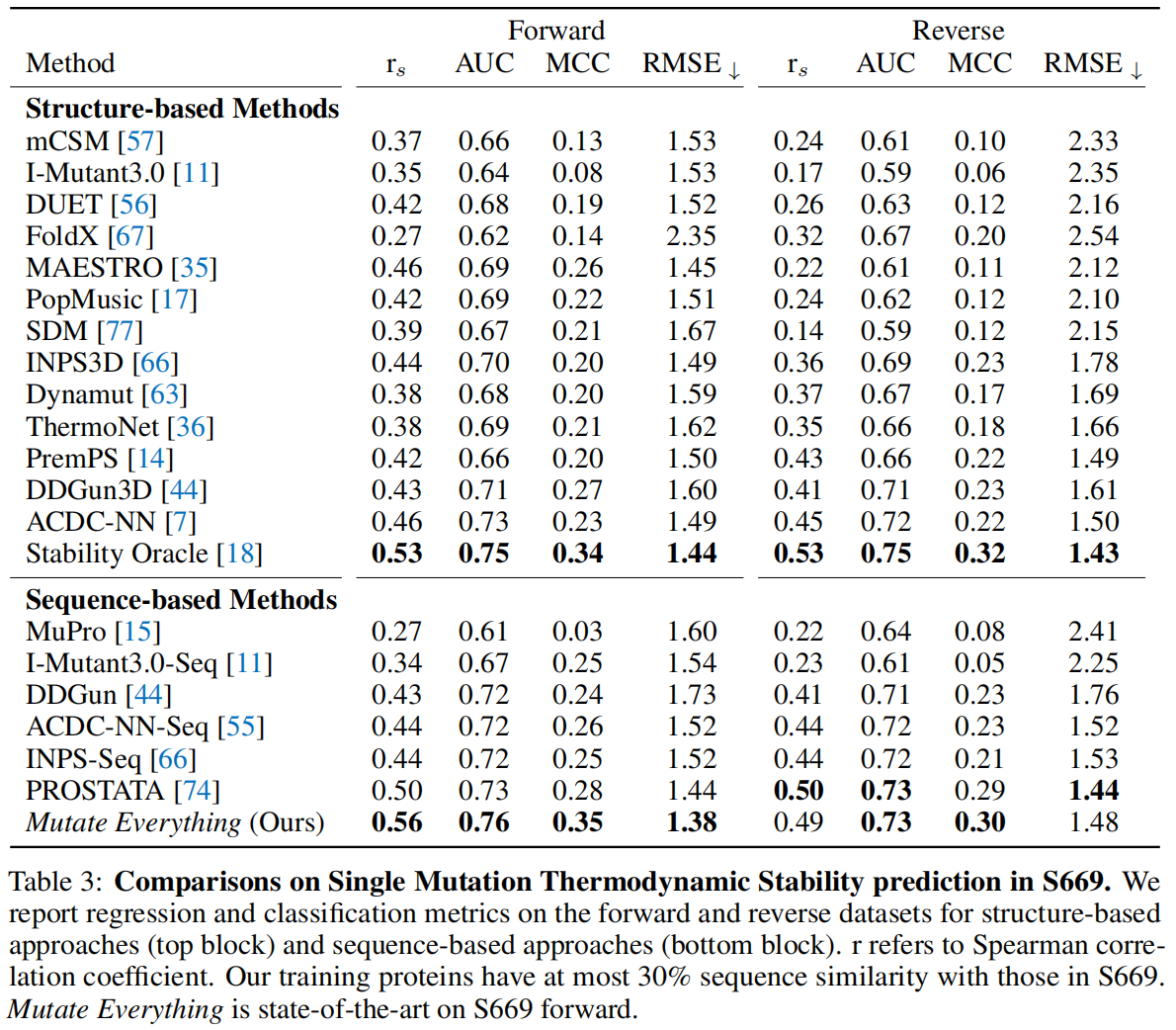
研究还验证了模型在预测单点突变下热力学稳定性变化的能力 。研究者们将Mutate Everything与先前的工作在最新引入的S669数据集上进行了比较 。S669数据集的建立旨在解决数据泄露问题,并为现有方法提供公平的比较平台 。同时,研究者们也评估了常见的反向数据集(reverse dataset)设定,即模型以突变后的序列作为输入,预测突变回野生型序列的ΔΔG值 。其对应的实验值是通过将从野生型到突变型的原始ΔΔG值取反得到的 。S669数据集经过精心策划且内容广泛,包含94个蛋白质,总计669个突变 。为了确保训练集和验证集的分离,S669中的蛋白质与训练集中的蛋白质同源性最高不超过30% 。
表3展示了与先前工作的比较结果 。Mutate Everything在S669上达到了0.56的Spearman相关系数,取得了SOTA性能,而之前的最佳水平为0.53
5.5 Generalization to other Phenotypes
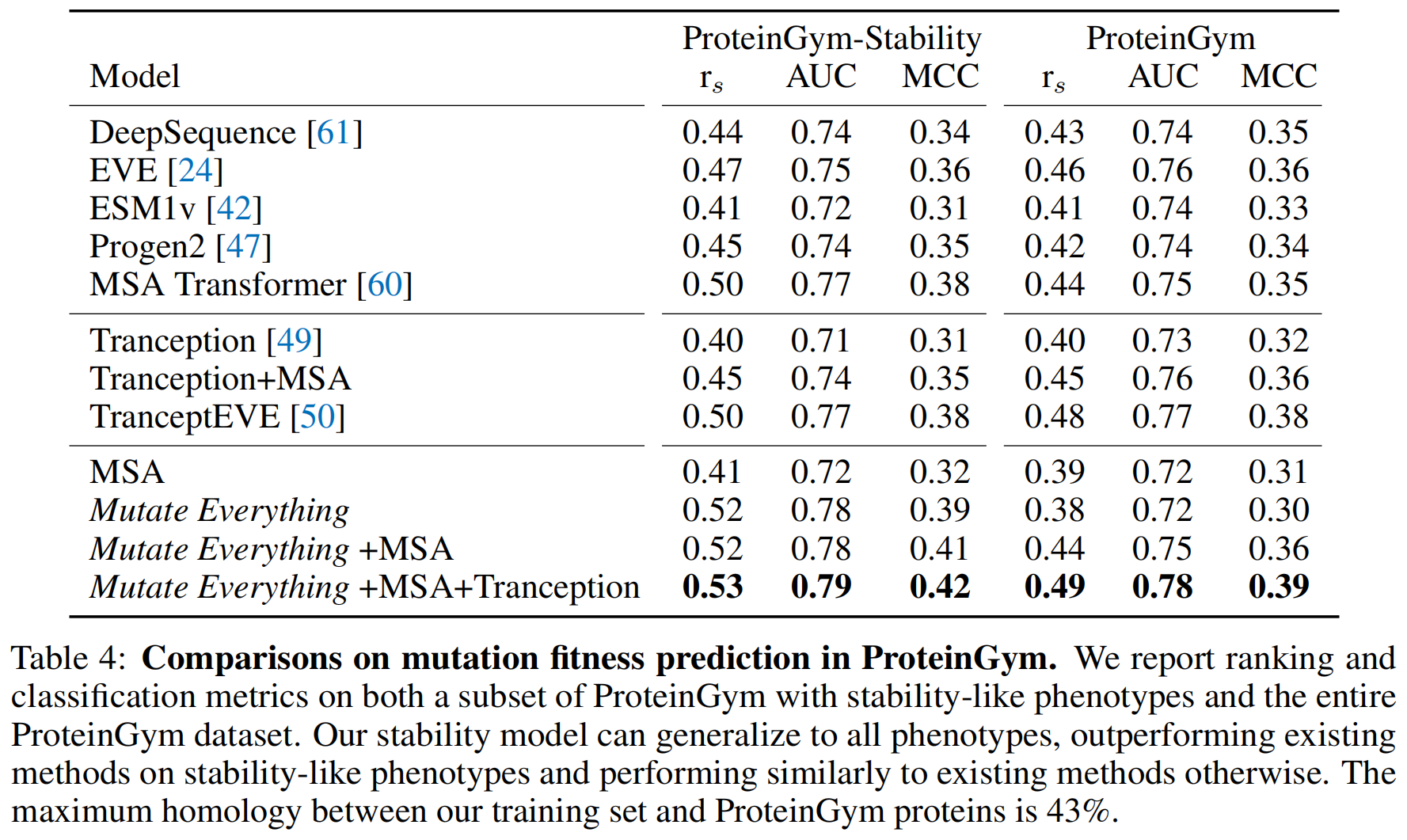
研究还表明,稳定性预测器也能泛化到其他表型的预测 。ProteinGym substitutions 是一个包含87个蛋白质、160万个突变集的数据集,这些突变集被标记了通用的适应度评分(fitness score),例如活性、生长速率或表达量等 。这些突变集最多包含20个替换,其中14个蛋白质含有高阶突变 。ProteinGym-Stability 分割则包含7个蛋白质和26,000个突变,其适应度标签已知与热力学稳定性相关(如热稳定性、丰度、表达量) 。ProteinGym中的蛋白质与训练集中的蛋白质序列相似性最高不超过45% 。由于内存限制,蛋白质被截断至最多2000个氨基酸
5.6 Ablation Studies
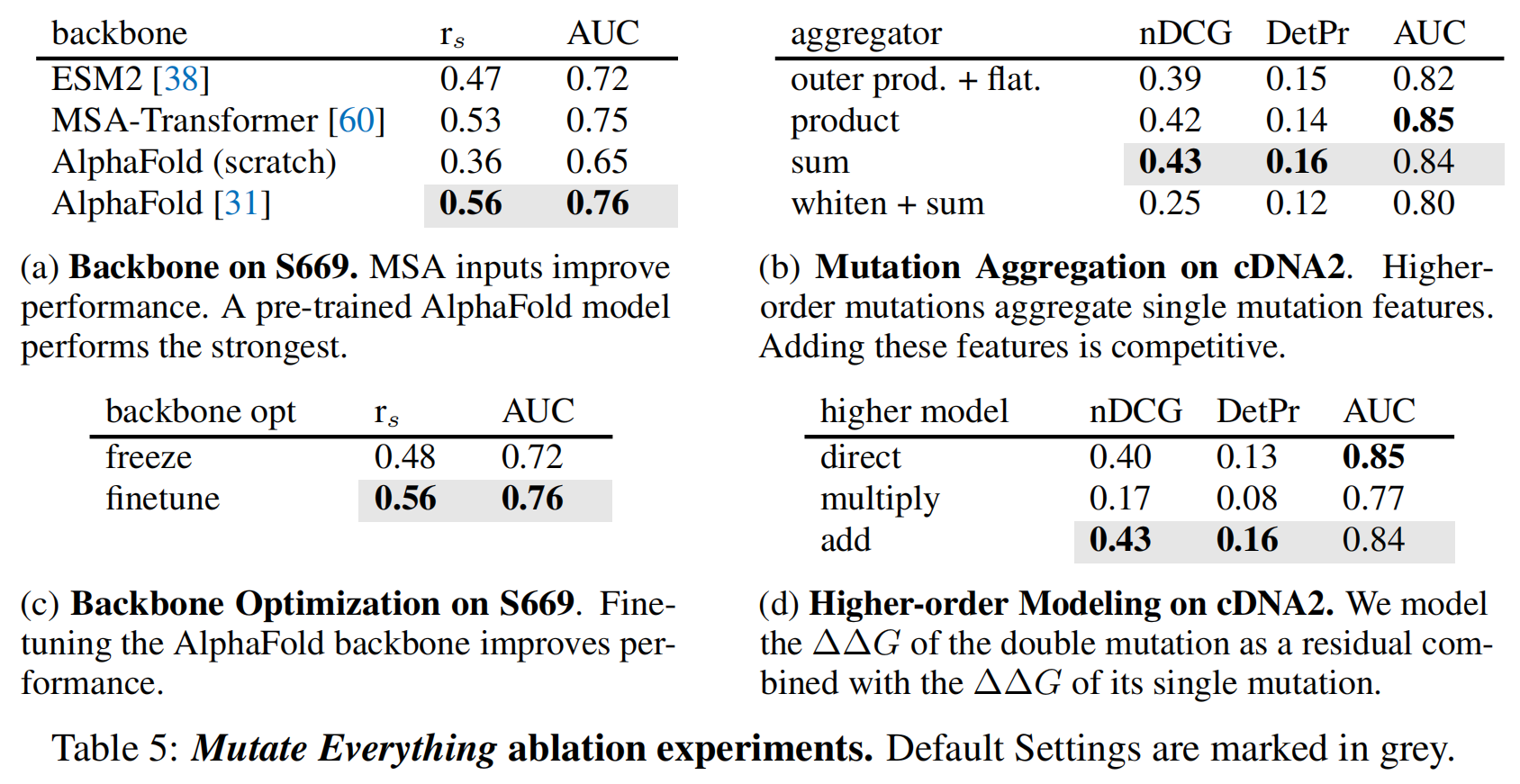
表5对几个架构设计选择进行了消融研究 。核心架构设计在S669上进行评估 ,而仅针对多重突变的架构设计则在cDNA2上进行评估 。
- 骨干网络(Backbone):表5a分析了不同特征提取器对单点突变稳定性评估的影响。ESM2仅以序列为输入,而MSA-Transformer和AlphaFold则同时以序列及其MSA为输入。ESM2和MSA-Transformer均在掩码残基建模任务上训练,而AlphaFold还在结构预测任务上训练。结果表明,MSA输入能提高稳定性预测性能,而AlphaFold的架构和训练能进一步提升稳定性预测。预训练的AlphaFold模型表现最佳。
- 骨干网络优化(Backbone Optimization):表5c比较了在稳定性微调过程中微调和冻结AlphaFold骨干网络的效果。结果发现,微调骨干网络能提高稳定性预测性能 。
- 聚合方式(Aggregation):表5b对聚合高阶突变表示的技术进行了消融研究 。为了控制特征维度大小,在使用外积并展平进行聚合时,头部的维度有所减小。在双突变上,使用乘积和求和进行聚合的表现相似。研究者选择了特征相加的方式,以便自然扩展到更多突变的情况 。
- 高阶建模(Higher Order Modeling):在直接预测模型中,多突变头部直接预测ΔΔG 。在乘法和加法模型中,多突变头部学习单个突变之间的相互作用如何影响高阶突变的ΔΔG;在这些模型中,多突变头部的输出学习的是对单点突变ΔΔG之和的加性偏置或乘性缩放 。表5d显示,在这三种选项中,学习一个偏置(即加法模型)表现最强 。

