论文地址:Mixture of Experts Enable Efficient and Effective Protein Understanding and Design
论文实现:https://huggingface.co/genbio-ai/AIDO.Protein-RAG-16B-proteingym-dms-zeroshot
AIDO.Protein:MOE预训练蛋白质语言模型
Abstract
蛋白质在生命中扮演着基础角色 ,理解其语言对于生物学和医学等领域至关重要 。尽管如ESM2-15B和xTrimoPGLM-100B这样的大型蛋白质语言模型(PLMs)表现优异,但它们的密集Transformer架构导致计算效率低下 。为了解决这个问题,本研究引入了AIDO.Protein ,这是蛋白质领域第一个专家混合(MoE)模型,拥有160亿参数 。它利用稀疏MoE架构,每个Transformer块有8个专家,并为每个输入token激活2个 ,从而显著提高了训练和推理效率。该模型在UniRef90和ColabfoldDB收集的1.2万亿氨基酸上进行了预训练 。AIDO.Protein在xTrimoPGLM基准测试的大多数任务中取得了SOTA结果 ,并在超过280个ProteinGym DMS分析中,达到了最佳MSA模型性能的近99%,且显著优于不使用MSA的先前SOTA模型 。此外,它在结构条件下的蛋白质序列生成任务中也取得了新的SOTA 。
1 Introduction
蛋白质作为生命的基本执行者,承担着细胞内大部分生物学功能 。它们的功能多种多样,包括作为生物催化剂、提供结构支持、运输分子、识别病原体以及传递信号等 。
为了理解蛋白质的功能,研究主要有两个方向:
- 序列-结构-功能: 通过研究蛋白质的3D结构(其活性形式)来理解其功能。
- 序列-功能: 直接从序列信息推断功能,尤其是在蛋白质结构信息稀缺的情况下。 无论哪个方向,都凸显了理解蛋白质序列“语言”的共同需求。
理解蛋白质语言对于推进遗传学研究和加速药物发现至关重要 。例如,它可以帮助设计能分解塑料垃圾或水解污染毒素的酶 ,或是在大流行期间及时创造疫苗 。
人工智能的介入:
- 大型语言模型的启发: 人工智能,特别是大型语言模型(LLMs)的最新进展,为实现这一目标提供了有前景的途径。LLMs在自然语言处理(NLP)领域的巨大成功,激发了研究者在蛋白质领域应用自监督预训练,即使用无标签的蛋白质序列进行训练。
- 蛋白质语言模型 (PLMs): 这些PLMs在蛋白质结构预测、功能预测和序列设计等多种任务中展现了卓越的性能。例如,基于PLM的ESMFold在结构预测上达到了原子级精度,接近AlphaFold2的水平,而拥有1000亿参数的xTrimoPGLM在多种功能预测任务上表现优越。
- 规模与效率的权衡: 模型规模是提升性能的关键驱动力之一,趋势表明更大的模型性能更好 。然而,随着PLMs变大,其计算效率会下降,因为运行更大模型需要更多计算资源 。现有工作主要关注通过扩大模型规模来提升性能,而较少关注计算效率 。
研究问题与解决方案:
- 核心问题: 能否在保持或提升PLM性能的同时,维持其训练和推理的效率?
- 潜在方案: 稀疏专家模型(Sparse expert models)提供了一个可能的解决方案。
- 稀疏专家模型机制: 在这些模型中,一部分参数被分为“专家”,每个专家有独立的权重。模型会将输入样本路由到特定的专家进行处理。因此,每个样本只与网络参数的一个子集交互,这与密集模型不同。由于只使用了一部分专家,计算量相对于总模型大小可以保持较小。
- 专家混合 (MoE): MoE是一种突出的稀疏专家模型,当集成到Transformer中时,它已成为NLP领域密集Transformer模型的有力竞争者 。
本文工作:
- 首个蛋白质MoE模型: 本文探索了在蛋白质领域预训练第一个MoE模型,这与所有现有的采用密集Transformer架构的PLMs不同。
- AIDO.Protein: 提出了AIDO.Protein,一个拥有160亿参数的MoE模型,在从UniRef90和ColabfoldDB收集的1.2万亿个氨基酸(token)上进行了预训练。
- 效率: 在训练和推理过程中,每个输入token由45亿参数处理,仅使用了总参数数量的28%。
- 评估: 在xTrimoPGLM基准(18项任务)和ProteinGym DMS基准(283项蛋白质适应性预测任务)等广泛任务上评估了模型。
- 结果: 实验结果显示,AIDO.Protein在保持更高计算效率的同时,在各项任务中均取得了强大的性能。它还被用于蛋白质逆向折叠,并超越了先前的SOTA方法。
- 结论: 这些结果表明AIDO.Protein是一个强大且高效的蛋白质理解与设计基础模型 。
3 Pre-training AIDO.Protein
3.1 Model architecture
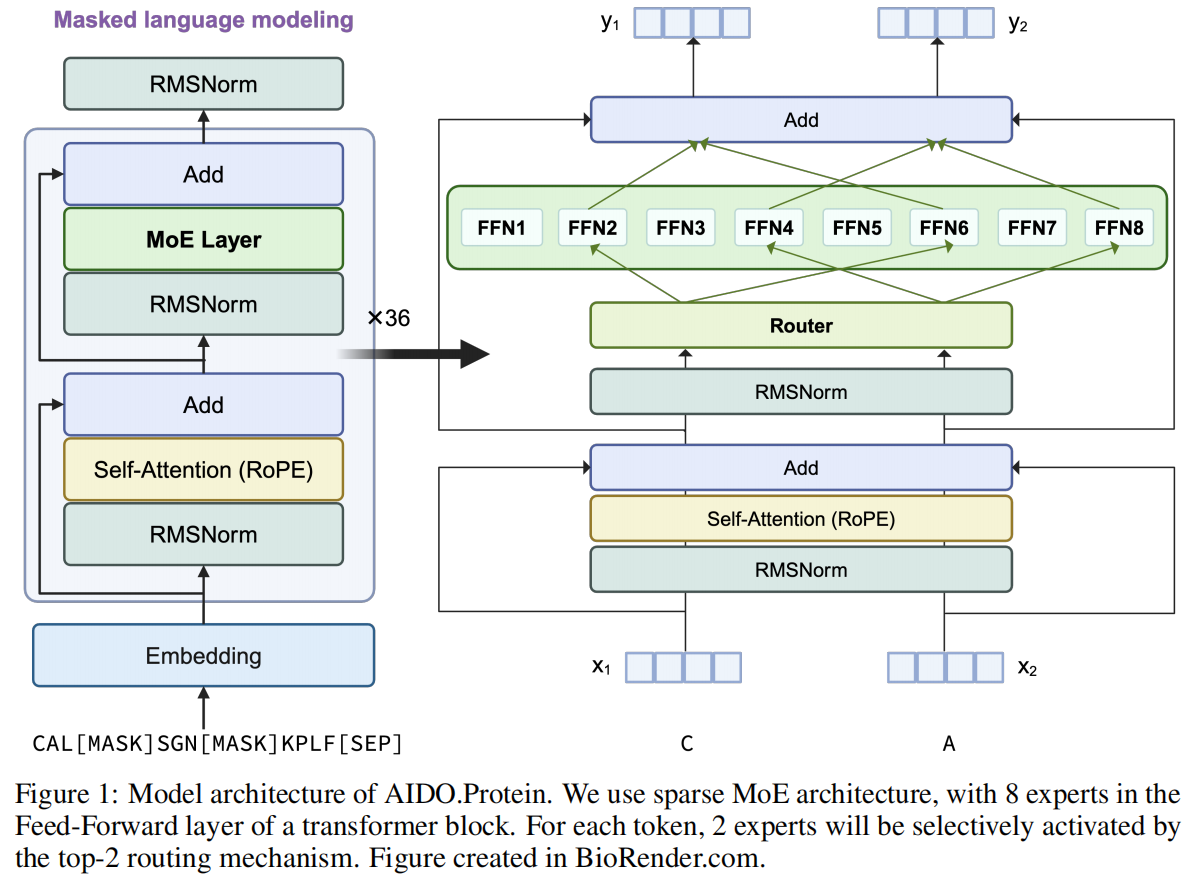
AIDO.Protein的模型架构基于Transformer编码器,但进行了关键修改以提高效率。
-
核心设计: 模型采用了Transformer编码器架构,但将每个Transformer块中的密集MLP(多层感知器)层替换为了稀疏MoE层。
-
MoE层:
-
其设计很大程度上遵循了Mixtral 8×7B 模型的MoE层。
-
这一层对输入序列中的每个token(氨基酸)独立应用。
-
它包含N=8个专家网络(Ei(x))和一个路由网络(Wg)。
-
对于每个输入token x,路由网络会计算权重,并通过TopK机制选择K=2个最相关的专家。
-
输出 y 是所选专家网络输出的加权和,其计算方式如下:
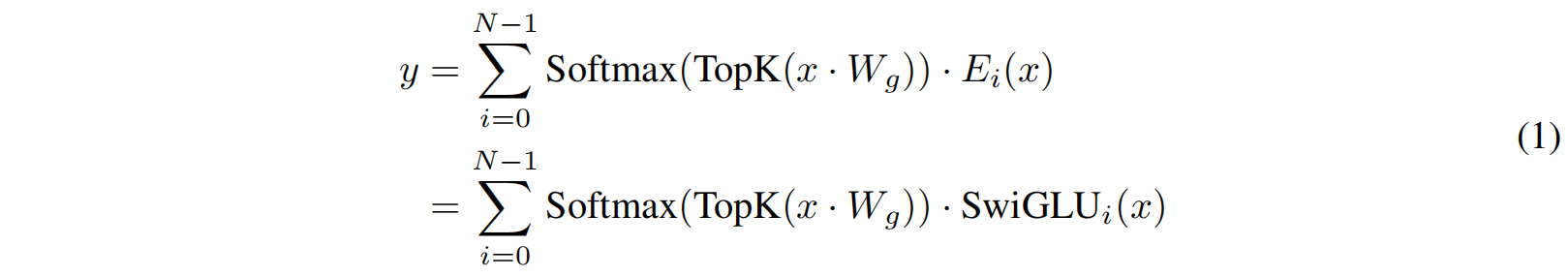
-
模型参数: 在实验中,设置 N=8, K=2, d=2304。模型包含36个Transformer层和36个注意力头,总参数达到160亿。
-
计算效率: 尽管总参数量巨大,但在训练和推理期间,每个输入token仅由45亿参数处理,这只占总参数量的28%。
-
训练目标: 使用标准**掩码语言建模(MLM)**目标进行训练。模型需要预测序列中被掩盖的氨基酸,从而学习蛋白质序列中固有的复杂依赖关系。MoE层有助于模型为不同类型的序列模式分配不同的专家,从而捕捉更广泛的序列特征。
-
可视化: 图1展示了该架构,包括带有RoPE(旋转位置编码)的自注意力层和包含路由器及8个前馈网络(FFN)的MoE层。
-
3.2 Pre-training data
为了训练AIDO.Protein,研究者们构建了一个大规模且经过精心筛选的蛋白质序列数据库。
- 数据来源: 模型最初在ColabfoldDB和UniRef90数据库的组合上进行预训练。
- ColabfoldDB: 侧重于宏基因组数据源,如BFD、MGnify以及各种真核和病毒数据集。
- UniRef90: 提供了来自UniProt知识库的聚类序列集,以全面的覆盖度和最小的数据冗余度为特点。具体使用了UniRef90/50(2022年12月之前的数据)。
- 持续训练: 鉴于UniRef90对先前PLMs的有效性以及持续训练对下游任务性能的提升作用,模型在UniRef90上额外训练了1000亿个token。
- 模型版本: 共开发了两个版本:
- AIDO.Protein-16B: 在ColabfoldDB和Uniref90的1.2万亿氨基酸上训练。
- AIDO.Protein-16B-v1: 在前一版本基础上,又在Uniref90上持续训练了1000亿氨基酸。
3.3 Pre-training settings
预训练过程采用了特定的超参数和多阶段策略。
- 通用设置:
- 全局批量大小为2048,上下文长度为2048。
- 优化器使用AdamW,权重衰减为0.1。
- 学习率调度采用余弦退火,预热比例为总训练步数的2.5%。
- 使用FP16混合精度训练以加速。
- 训练框架为Megatron-Deepspeed,使用了256块Nvidia A100-80G GPU,耗时25天。
- 三阶段训练:
- 第一阶段: 在UniRef90和Colab数据库的1万亿token上训练18.5天,学习率从2e-4衰减到2e-6。
- 第二阶段: 在相同数据源的2000亿token上训练4天,学习率从1e-5降至1e-6。
- 第三阶段: 在UniRef90的1000亿token上进一步训练2.5天,学习率在1e−5到1e-6之间。
4 Experiments
4.1 AIDO.Protein achieves strong results across diverse tasks from xTrimoPGLM benchmark
为了全面测试模型在各种蛋白质理解任务中的能力,研究人员在xTrimoPGLM基准上进行了评估 。该基准包含18项不同的任务,分为四类 :
- 蛋白质结构预测: 接触图预测 (Contact map prediction),蛋白质折叠预测 (Fold prediction),二级结构预测 (Secondary structure prediction)
- 蛋白质功能预测: 抗生素抗性预测 (Antibiotic resistance prediction),荧光预测 (Fluorescence prediction),适应性预测 (Fitness prediction),定位预测 (Localization prediction)
- 蛋白质-蛋白质相互作用预测: 酶催化效率 (Enzyme catalytic efficiency),肽-HLA/MHC亲和力 (Peptide-HLA/MHC affinity),金属离子结合 (Metal ion binding),TCR-pMHC亲和力 (TCR pMHC affinity)
- 蛋白质开发: 溶解度 (Solubility),稳定性 (Stability),温度稳定性 (Temperature stability),最适温度 (Optimal temperature),最适pH (Optimal ph),克隆分类 (Cloning clf),材料生产 (Material production)
微调方法: 所有任务都使用了**LoRA(Low-Rank Adaptation)**进行微调,设置秩(rank)为16,alpha为16
优化器与学习率:
- 大多数任务使用了Adam优化器,峰值学习率为1e-4,并配合余弦学习率调度器(预热比例为0.05)。
- 对于接触图预测任务,则使用了Adam优化器和1e-4的恒定学习率 。
训练周期与选择: 模型被微调了10、15或20个周期(epochs),并根据验证集上的得分选择最佳的模型检查点
4.2 AIDO.Protein demonstrates impressive performance on ProteinGym DMS benchmark
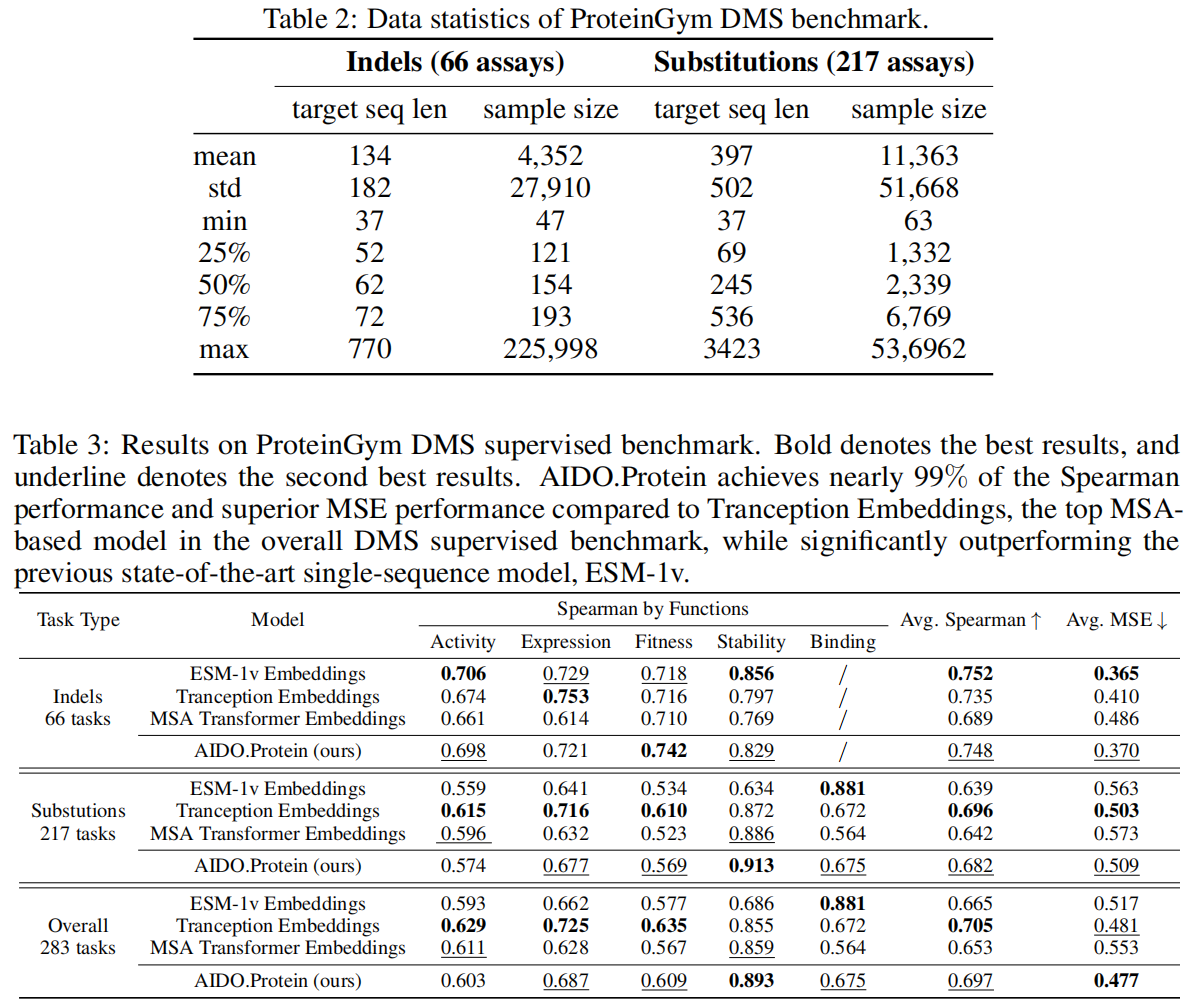
该基准测试包含66个插入/删除(indels)分析和217个替换(substitutions)分析 。每个分析都提供了针对特定目标蛋白质的所有可能突变及其相应的适应性得分 。评估指标采用Spearman秩相关系数和均方误差(MSE)
4.2.1 DMS indels supervised benchmark
微调设置:
- 所有任务均在5折交叉验证(ProteinGym提供的随机划分方案)下评估。
- 对于54个小样本任务,使用线性探测(Linear Probing),即冻结AIDO.Protein的参数以减轻过拟合。预测头是一个2层MLP(隐藏层大小128,dropout率0.1)。使用AdamW优化器,峰值学习率1e-3,余弦学习率调度(预热比例0.05)。不使用验证集进行模型选择,而是直接训练1000步,并使用最后一个检查点进行预测。
- 对于其他12个大样本任务,使用LoRA微调(rank 16, alpha 32),峰值学习率1e-4 。使用一个折作为验证集,训练10,000步并采用早停策略。
4.2.2 DMS substitutions supervised benchmark
任务描述: 替换突变是指将蛋白质序列中的一个或多个残基替换为其他残基。该基准包含217个分析,其中69个为单点替换,148个为多点替换。
数据特点: 如表2所示,序列长度从37到3423不等,样本量从63到536,962不等。小样本任务易过拟合,而超长序列会导致内存溢出(OOM)问题。因此,根据样本大小和序列长度采用了不同的微调策略。
模型选择: 评估了AIDO.Protein-16B-v1版本,因为它在Uniref90上的持续预训练显著提升了零样本替换预测性能。
微调设置:
- 采用随机5折交叉验证方案。
- 使用Adam优化器和余弦学习率调度。
- 使用LoRA微调(rank 16, alpha 32),峰值学习率1e-4,训练10,000步,并设有早停机制。
- 对于样本量超过20,000的13个任务,每个折只训练一个epoch。
- 对于序列长度超过2048的4个任务,将序列截断至2048,并调整LoRA rank为4,alpha为8。
4.3 DMS overall supervised benchmark
在表3 评估了插入/删除(Indels)和替换(Substitutions)任务的性能之后,研究人员进一步考察了 AIDO.Protein 在整个 DMS 监督基准上的整体表现。这部分综合了 66 个 Indels 分析和 217 个 Substitutions 分析的结果,共计 283 个任务。
4.4 AIDO.Protein offers enhanced capabilities for protein design

蛋白质设计在生物化学、分子生物学和生物技术领域是一项至关重要的研究和应用。本节讨论了如何将 AIDO.Protein 模型应用于这一目的。
核心应用:蛋白质逆向折叠 (Protein Inverse Folding)
- 研究团队特别关注了蛋白质逆向折叠,这是从头设计蛋白质(de novo protein design)的关键步骤。其目标是生成能够折叠成特定三维结构的蛋白质序列。
- 他们为此开发了一个基于离散扩散模型 (discrete diffusion modeling) 的框架。
框架细节与方法:
- 结构编码器: 该框架使用了 ProteinMPNN-CMLM (ProteinMPNN 的一个变体) 作为结构编码器,以实现结构条件下的扩散过程。
- 详细信息: 关于该框架的更多技术细节可以在论文的附录 A.1 中找到。
实验与评估:
- 数据集: 实验使用了 CATH 4.2 数据集进行评估,这是一个公认的蛋白质设计评估资源。
- 结果 (见表 4):
- 该框架被命名为 AIDO.Protein-IFdiff。
- 它取得了 58.26% 的中位序列恢复率 (Median Sequence Recovery)。
- 这一结果显著高于之前所有的最先进(SOTA)方法,例如 LM-Design (54.41%) 和 DPLM (54.54%)。
示例展示 (见图 2):
- 论文展示了一个具体的例子(PDB ID: 2DLX, Chain: A)。
- 图中比较了:
- 左侧: 使用 AIDO.Protein-IFdiff 框架生成的序列,以及通过 AlphaFold2 预测的该序列的 3D 结构(并用 pIDDT 值显示了预测置信度)。
- 右侧: 来自蛋白质数据库 (PDB) 的真实(Ground truth)序列和结构。
- 这个例子显示,生成的序列所预测的结构与真实结构高度吻合,并且预测置信度很高,证明了该框架在生成有效蛋白质序列方面的能力。

